Sphincters are beginning to loosen in LA with the MMA fiasco tapering into a more banal conclusion. Here MMA does not refer to Mixed Martial Arts. It is the abbreviation for methyl methacrylate, a reactive monomer for the production of PMMA, poly[methyl methacrylate], often called acrylic or Plexiglass. Plexiglass is a generic name like “Xerox” or “Velcro” but it is also trademark.
There was a lot of loose talk about a possible explosion, a B.L.E.V.E., detonation, and vapor plumes. I would categorize this incident as a low frequency, high consequence event.
This being LA, I wonder how long it will take for a movie or miniseries to come out? I vote for Brad Pitt and Scarlett Johansson to star in it and produced by Steven Spielberg. The lead investigator for the US Chemical Safety Board would be Tig Notaro.
Google Earth view of GKN Aerospace location, 12369 Western Ave, Garden Grove, California. Site of evolving chemical (MMA) runaway incident. The three smaller silver-colored tanks are presently being sprayed down with cooling water.
Cooling a large tank of liquid on the outside surface is one of the least effective methods of cooling bulk material. Ideally, a vessel would have an immersed heat exchanger for efficient cooling with agitation and possibly with a cooling jacket. Properties of MMA: 100.3 to 101 oC boiling point; Flash Point 10 oC; vapor pressure of 29 to 37 mm Hg, It is highly flammable.
Google Earth view of GKN Aerospace location, 12369 Western Ave, Garden Grove, California, site of evolving chemical (MMA) runaway incident. To the right of the large tank are the three smaller tanks presently being sprayed down for cooling. Notice the proximity of a residential neighborhood.
It is not uncommon for an industrial park to be near a residential area. But, in my estimation, a chemical plant storing something like bulk MMA near a residential area is not the best situation. You have to ask if the zoning people knew about this proximity situation or if such was even contemplated. This is likely to lead to state legislation or city ordinances. Everyone likes to rush into locking the barn door after the horse has run loose.
Google street view of 12369 Western Ave, Garden Grove, California. Site of evolving chemical runaway incident. The tanks presently being sprayed down for cooling are behind the brown and white tank.
According to Wikipedia, GKN Ltd is a British multinational automotive and aerospace components business headquartered in Redditch, England. They manufacture what are called military “transparency systems”, meaning canopies, windshields and windows.
The MMA in the tanks is converting to a big block of acrylic polymer. If you’re interested in buying it, I’ll bet they will give you a good deal on it …
The methyl methacrylate (MMA) runaway situation in Garden Grove, CA, seems to have plateaued. The aerospace company whose storage tanks are in the news produces what it calls “transparencies”. This is their trade name for plexiglass aircraft canopies, wind shields and windows. The word “plexiglass” is in common usage but derives from the trademark “Plexiglass“. Its not to be confused with “Lexan” which is a polycarbonate.
It dawned on me that the MMA runaway situation will be self-limiting. As the runaway polymerization proceeds, the rate of heat evolution should taper off as the reaction rate slows due to consumption of MMA. Just simple kinetics. The runaway reaction converts liquid MMA to solid poly[methyl methacrylate], or just PMMA. The MMA is turning onto a solid brick of plastic. This slows down the rate of reaction which necessarily slows down the rate of heat release.
The upshot is that if the tanks are kept cool enough to prevent rupture, yet warm enough to allow the reaction to creep forward, the runaway is under control.
The very fact that this runaway happened at all in “normal” storage suggests that the MMA was insufficiently passivated with stabilizer. The usual stabilizers are typically some variety of phenolic additives. This would include BHT, BHA and other catechols and phenols chemically modified for organic solubility.
The polymerization of MMA occurs via a radical chain reaction mechanism. Stabilizers like the phenolics are able to release H-dot, or hydrogen radical, which interferes with the polymerization by terminating the free radicals propagating the reaction. Each H-dot combines with any other radical it comes in contact with and halts MMA participation in the reaction.
Below is a graphic from an article published on PubMed. Watch the dots, the dots are the radical electron. The upper reaction is chain propagation which is the desired reaction. See how the double line in the MMA structures changes to a single line? One of the two electrons in the second line is taken by an incoming free radical to form a new link in the growing polymer. The other electron is the leftover free radical which awaits a collision with another MMA molecule. This is how the polymer grows.
Image Source: Hint- watch the dots. Ahamad Said MN, Hasbullah NA, Rosdi MRH, Musa MS, Rusli A, Ariffin A, Shafiq MD. Polymerization and Applications of Poly(methyl methacrylate)-Graphene Oxide Nanocomposites: A Review. ACS Omega. 2022 Dec 15;7(51):47490-47503. doi: 10.1021/acsomega.2c04483. PMID: 36591191; PMCID: PMC9798503.
Let’s assume that the first reaction is the desired reaction and I’ll refer to it as “proper”. The critical feature of MMA is the double line. That is the location of the transformations in the polymerization. Each double line in the MMA has two ends- one end is the terminus and the other end is internal. The addition of the next MMA is desired to add its terminus of the growing end of the chain. This transformation leaves a radical at the internal end for further desired polymerization.
The second reaction shows how propagation can run afoul when the double line connects at the wrong position. The shape of this connection error- or kink- is different than that of the intended connection. The result is a polymer backbone linkage of 2 carbon atoms rather than the desired single carbon. This doesn’t happen just once but many times. The cumulative effect is that a chemically distinct polymer is produced that degrades or dilutes the desired physicochemical properties of the intended PMMA.
The third and lower reaction shows the undesired linkage reaction with both “properly” linked MMA and “improperly” linked chains to give a succeeding undesired linkage.
Note: “Desired” and “undesired” refer to intended connectivity and the unintended, kinked, connectivity. In reality, the desirability of any polymerization process is determined by the resulting set of physicochemical properties of the bulk polymer. Knobs are twisted, levers pulled and temperatures set in the plant to give the desired product with its combination of linkages.
I wonder if during the event that if a trickle of stabilizer were added, would it have made an observable difference? I assume that no agitation in the tank is available beyond convection currents, of course. it would be a large scale science experiment on an evolving and novel public emergency situation. Responders and, uh, everyone else, would disapprove.
According to a March 29, 2026, report in OilPrice.com theft in the Permian Basin of Texas amounts to 1 to 2$ billion annually. The Permian Basin, source of benchmark West Texas Intermediate (WTI) crude, reportedly holds 15 % of the global oil reserves. Oil theft is nothing new but recent high prices for oil have increased theft. Anything not bolted down like copper or tools at a well site will likely be stolen. This is a world-wide problem.
Source: USGS. Red is gas and green is crude oil. The dark outline is the Permian Basin area.
In the above USGS map the perimeter of the Permian Basin is shown as the heavy black line. The Permian Basin is up to 25,000 ft thick and is a sedimentary formation that dips to the eastward.
Source: Wikimedia. The Permian Basin has several sub-basins with the Midland and the Delaware being the largest.
According to Bloomberg (paywall) producers are losing between $1 and $2 billion per year. Martin County Sheriff Randy Cozart estimates 500 barrels of crude oil are stolen each week. A common method of stealing the oil uses a vacuum truck siphon out the crude from tanks on the well site. Some thieves use a waste hauling truck which is normally present in the oil patch.
Naturally, the caped crusaders at the Texas Railroad Commission are none too pleased about the thefts. According to one source, a truckload of 180 barrels of crude was worth about $15,000. The stolen oil could go into the tanks of another producer or directly to the gathering facility for transfer into a pipeline.
In my industrial career as a PhD organic/organometallic chemist I was kept busy for about 10 years with in-house Reaction Calorimetry (RC), Accelerating Rate Calorimetry (ARC), Differential Scanning Calorimetry (DSC) as well as Thermogravimetric Analysis (TGA) for validating thermal process safety. To institutionalize this I was asked to start a process safety department and began standardizing experimental protocols and a database for the results. I was able to scour the internet for thermochemical papers, looking for mentions of energetic properties. As always, much can be learned by just looking around.
Thermal process safety refers to safe operation of chemical manufacturing in regard to the generation of heat in a reaction mass and the hazards arising therein. The hazards from uncontrolled self-heating include acceleration of reaction kinetics producing accelerating heat and pressure evolution. If the reaction enthalpy and subsequent temperature rise theoretically exceeds the boiling point of the solvent despite the cooling jacket and the chilled condenser, then self-heating can lead to a boil up and uncontrolled ejection of the reaction mass. With insufficient cooling, the temperature will rise to the solvent bp and boil off the solvent first, carrying much heat away as heat of evaporation. Once most of the solvent has boiled away and if the reaction mass continues to self-heat, the temperature will continue to rise and peak at some undesired level as the reactants are consumed. Further heating of the now hot, highly concentrated reaction mass, potentially leading to successive reactions that may or may not be exothermic.
When a liquid phase reaction mass self-heats faster than heat can be removed, the reactor pressure will begin to rise. As pressure builds, the boiling point of the reaction mass begins to rise, slowing down the boil-off. A sudden drop in pressure, as with the burst of a rupture disk, will cause a superheated solution to promptly boil throughout the reaction mass. This means that flash vaporization can lead to bubble formation throughout the volume of the reaction mass producing a foam. The severity will depend on the pressure drop and the bp of the solvent. If the headspace is sufficiently small, the foam can expand rapidly and begin to exit through the vent pipe. A properly engineered vent pipe has been sized to vent gas/vapor at specified conditions. Since a foam is part liquid and part gas/vapor, it lacks the overall compressibility of a gas/vapor so the resulting foam flow may be lower than calculated for a gas/vapor, slowing the rate of depressurization.
The distinction between gas and a vapor is that a vapor may be condensable as with most solvent vapors, but evolved gases like hydrogen, methane or carbon dioxide will combine with a non-condensable blanket gas like nitrogen and resist condensation in the by the chiller. The point is that if one is relying on a chilled condenser to knock down non-condensable gases as a pressure management control, then a rude shock is headed your way, especially if the rupture disk bursting pressure is higher than need be.
Flow into vent pipes that exit outdoors may discharge hot reaction mass onto the roof or wherever the vent terminates. If the vent terminates into a knockdown drum or other catch vessel, the hot reaction mass contacts whatever may be in those vessels.
Image: Mettler Toledo. The Mettler-Toledo RC1 rigged for dual feed and distillation or reflux. The two brown bottles (lower right) sit on balances and feed from two reagent bottles into the reactor (lower left). The feed of liquid reactants is pre-programmed and is controlled quite accurately. Reagents are fed into an agitating reaction mass (yellow) while the temperature and enthalpy (H or h) are monitored on the fly. The instrument monitors the jacket and reactor temperatures and with the help of heat capacities, Cp, can display the enthalpy of the reaction as it proceeds.
Fortunately, the thermal profile leading up to the above scenario can be modeled in properly conducted RC1 experiments. But exactly what can be done beforehand?
First, let’s realize that the total self-heating temperature rise can be measured. We add that ΔT (temperature rise) to the proposed reaction temperature Tr and get a maximum temperature of the synthetic reaction, MTSR. Once we have this, if the MTSR is greater than the bp of the solvent(s), then we know that an uncontained runaway is possible. What to do then?
R&D needs to justify the problematic low boiling solvent with the reaction temperature to be applied.
R&D needs to provide input on lowering the design reaction temperature.
Is a lower Tr for a longer reaction time feasible?
How sensitive is the reaction to a higher bp solvent substitution?
If the chosen solvent conveniently forces side or waste products to precipitate and be removed by filtration, then we have the conundrum of safety vs efficient processability.
The magnitude of the hazard in the minds of everyone involved may be quite different and will require a documented decision process. Engineering input here is invaluable.
Thermal runaway profile. Source. The linked article is well written.
Tp Process Temperature
ΔTad Adiabatic Temperature rise
MTSR Maximum Temperature of the Synthetic Reaction
TMRad Adiabatic Time to Maximum Rate
Tx Time of Cooling Loss
What if the design solvent is truly required for a feasible and economic process? This needs to be discussed first with chemists and engineers in the room and a decision rendered. If chemist input says no solvent change is desirable or economic, the engineers need to speak up as to whether there is an engineering work around. If a ready engineering work around is not feasible and the product is still important, then the chemists need to be challenged to find procedure that denies the equipment a runaway condition.
If we’re lucky, a partial batch reaction method can be used wherein the reactor is charged with solvent and most of the reactant compounds are in the vessel at the beginning of the run. The final reactant is slowly fed into the reactor and the reaction temperature is controlled by the feed rate. Reaction calorimetry can be used to arrive at a plausible maximum feed rate that is fast but not too fast. A reaction calorimeter is basically a chemical reaction detector and can be used to look for an approximate reaction onset temperature. Remember that onset temperature is not a physical or chemical property. It depends on the detection equipment and the rate of heating.
Like everything else, success can depend on first asking the right questions. On the graphic computer display of the RC1, you can determine the response to an aliquot of reagent addition. Does the heat production, q, rise promptly with addition or does it lag? If there is a lag or a latency, it means that over-charging by operators at scale can happen if they are looking for a prompt “heat kick” on addition of the feed.
The RC1 can also show the length of time to react away the feed or what the total reaction time may be If the reaction has a natural response lag, then a defined charge mass is called for. A response lag may also be due to the presence of water which must first be quenched under the reaction conditions. The most insidious situation is when the feed reactant accumulates in the reactor over the course of the reaction. This is very difficult to judge by the operators. The feed may accumulate until the reaction suddenly begins and accelerates out of control. This is not uncommon.
Finally, a proper “batch reaction” is one in which all of the reactants are loaded into the reactor all at once, the temperature is adjusted and the reaction begins. It is critical that before a new batch reaction is allowed, the chemists must show that this will not result in a runaway condition. This is where reaction calorimetry shines. The safety of a batch reaction is reproduced in the RC1and the progress is monitored. The RC1 can also be used to explore various reaction conditions to see if runaway potential can be easily blundered into. How narrow are the safe operating parameters? Many plant incidents happen at shift changes where the continuity of watchfulness may diverge for a time, even with automation.
TMRad Adiabatic Time to Maximum Rate
A very informative piece of data to have is the TMR- Time to Maximum Rate. This can be obtained by an Accelerated Rate Calorimeter or ARC. The instrument consists of a furnace into which is placed a sample “can” which can be made of metal or glass. The furnace raises the sample temperature gradually using a heat-wait-search (HWS) method searching for an onset temperature.
Once an onset temperature is found, the HWS is automatically halted and the furnace keeps adjusting its temperature to match the rising internal sample temperature. If the internal sample temperature and the exterior furnace temperature are the same, then the sample is under adiabatic conditions and no heat flows in or out of the sample can. The sample temperature is driven by self heating only.
Knowing the sample mass and the best guess at Cp, constant pressure heat capacity, the reaction enthalpy can be determined. From the data, the Time to Maximum Rate (TMR) can be calculated to give an equation. It is the time that a substance that is self-reacting takes to reach the maximum rate of heat output as a function of sample temperature. The instrument also records sample pressure. If the sample pressure does not return to ambient pressure at room temperature, this would mean that a non-condensable gas was evolved.
Image of the Phi-Tec II ARC system. from H.E.L. company. My ARC experience is with this model.
A typical ARC experiment took me from 6 to 24 hours to complete the HWS routine.
What TMR data allows one to do is to find a reaction temperature the reaches maximum rate in 24 hours or more. You plug in a temperature and you get a TMR. The temperature needed to produce a TMR of 24 hours is considered by many in industry to be the uppermost safe processing temperature. It helps to answer what the maximum safe temperature in which a solid can be dried before decomposition begins.
To outsource safety testing or not
First and foremost, a commercial safety test lab understands and uses procedures that are agreed upon and standardized. Also, if down the road there comes a related event, your response to criticism will be to refer to the test lab experts, not some ham fisted employee monkeying around in the lab doing improvised experiments. Certain safety matters should be referred to the commercial lab experts for valid results and for CYA. This applies especially to energetic materials like nitroaromatics or nitrate esters.
Chemical manufacturing is conducted at many scales from laboratory gram scale products for R&D, multi-kilogram kilo-lab batch processing to the colossal commodity scale continuous manufacturing of petrochemicals, agrichemicals, polymers, flavors & fragrances, and pharmaceuticals. Nearly all of these commodity chemicals and polymers are well known and have safety issues related only to flammability, exposure and dose.
Outsourcing tests that can be done inhouse is a missed opportunity to accumulate more skills which is company treasure. I’m speaking of calorimetry. Calorimeters can be brought on-site and meshed in with research and development. Just learning how to interpret thermograms alone brings workers new insights into their chemistry.
What is best for your company? In-house safety testing or outsourced safety testing? Like nearly everything else in life, the answer depends on the situation. If you need to survey for explosive hazards for the first time, there are several competent commercial labs available that will use standard protocols. My experience is that they employ just engineers or a mix of chemists and engineers. They conduct standard testing protocols wherein a series of samples are exposed step-wise to a series of ever increasing stimuli intensity to find the boundary conditions of sensitivity to various stimuli, like heat, friction, impact, dust explosion parameters, burn tests, static charge lifetimes and minimum ignition energy (MIE) with electrostatic discharge.
Explosibility testing
Sensitivity to explosive behavior is tested in numerous ways to flesh out the sensitivity profile. Testing is performed in stages where the least intense stimuli are tried first to screen for highly sensitive substances. The results of any single test run are graded as ‘Go/No Go’ or ‘positive/negative’. The terms ‘Go’ or ‘Negative’ mean that an explosive property was observed.
Part of explosives testing is finding out what kinds of stimuli lead to initiation of an explosion. The Bureau of Mines (BOM) drop weight test looks for the maximum safe impact energy. There is a friction test, an electrostatic discharge test, and many others. If the sample does not give a Go result at the maximum machine impact or friction, then it is regarded as safe under those precise conditions. In the BOM test, the higher the number (in drop distance of a 5 or 10 kg weight), the more stable it is to impact.
You get the testing data. Now what?
Now how do you take numerical test data and convert it to safer operations? This is where engineers can be most useful. Imagine a substance that has a 34 inch BOM drop weight result with a 10 kg anvil. Will any process equipment mash down on the substance inadvertently? Put this ball in the court of engineers and let them chew on it. This data moves workers closer to confidence in safety.
Outsourcing safety testing and explosive screening can lead to a conundrum. Outsourcing anything means that certain expertise may not be internalized for your company’s use, the user or manufacturer. Commercial labs will absolutely not comment on how the material can be safely used, whether or not it is too dangerous or nominally safe under your use conditions. Safe use is not an endorsement they will make, they will only stand behind their results from standard testing protocols. I’d do the same.
Before safety testing you were alone. Now, with safety data, you are still alone but with numbers. Engineers and plant operators are invaluable in locating equipment that delivers impacts or friction. They can also help to identify non-grounded equipment that may generate or accumulate electrostatic charge. Always get the plant people involved.
It didn’t take long to realize that if we sent samples out to commercial labs for calorimetry testing, the samples were subjected to unfamiliar standard test methodology. Early on it was fascinating to see what kind of experimental setups were used and what the results looked like. Being a synthesis chemist I was unfamiliar with calorimetry. My earlier exposure to calorimetry was limited to what appeared in molecular dynamics and mechanics modeling. Acquiring actual data on reaction enthalpies and onset conditions myself awakened a fascination that carried me far into reaction calorimetry and thermochemistry.
What was not clear at the outset of receiving external calorimetric, electrostatic and explosive test data was what to do with it. Using external hazard data to inform operational procedure was new to everyone. Yes, we could learn from an ARC experiment what temperature the onset to a runaway condition begins, but how to use the measurements in practice wasn’t always obvious.
Incidents have three phases- initiation, propagation and termination. You have to ask this: if an incident initiates, what is the preferred propagation direction to termination? Yes, this can be controlled somewhat but only in advance. For instance, if an explosion happens, what is the least terrible direction for the blast to go? These matters should be considered in the design phase of construction of a chemical facility. If they weren’t, then decisions must be made despite the lack of preplanning.
As an example, a commercial explosives company I’m aware of built their manufacturing facility out in the European countryside. Explosive materials were prepared, stored and handled in small buildings distributed over a large area with distance, berms and trees separating them. If an explosion happened, the blast wave would be isolated from other assets and attenuated by distance, berms and forest. Here, the propagation phase was suppressed by distance and topography.
Another explosion highlights the folly of not segregating manufacturing operations. A plant manufacturing a hydroxylamine called HOBT suffered a catastrophic incident where a reactor blew apart explosively during a process previously performed many times. The reactor was housed in a structure that had expanded over time by adding manufacturing space by piecemeal addition as needed. This resulted in a building that was a rabbits warren of rooms and hallways even including admin space. The explosion did not just happen without warning. The reactor began to overheat from accumulating heat of reaction and became unresponsive to cooling efforts by the operator. As the operator turned to go get help, the reactor exploded sending parts up and out of the building, with the agitator landing on the roof of an adjacent business and onto railroad tracks. Heat transfer oil ran out of the building and flowing into the nearby river. The operator was blown through a sheet rock wall but survived. The shock wave propagated into adjacent spaces and down hallways, blowing out windows, internal and external doors including overhead doors.
The sad thing is that another plant suffered a devastating explosion 20 years earlier making the same hydroxylamine product. Perhaps lessons were learned at this plant, but those lessons didn’t to the other plant.
The lesson is clear. In chemical manufacture the R&D folks must be sure that all chemical properties are well understood and such knowledge is a part of accessible in-house expertise. If there is no R&D, meaning that a large scale procedure is simply written up and performed without the scrutiny of cold expert eyes evaluating it, then you are stepping onto a high wire without a net. Both plants making the hydroxylamine had experienced chemists on site and performed the procedure without incident many, many times. Even then, incidents happened but how many incidents were averted by expert judgement? We’ll never know.
Experience
Let’s talk about experience. Career chemists are like everyone else- they may have accumulated years of experience. Some of the learning’s a person has accumulated are captured in writing and available to staff. Other learning’s reside in a person’s head only and are perhaps regarded as ‘obvious’. Or the serious hazards are actually disclosed on the Safety Data Sheet which was filed away without scrutiny. Knowledge of explosibility of a particular substance could be too narrow by virtue of time and obscurity to serve as walking around knowledge by many chemists. Some of us are accustomed to spotting explosive functional groups (explosophores) on a molecule but many are not.
For some individuals, their 18 years of experience is better described as 6 years repeated twice, or worse. Years of experience should always imply years of continuous improvement.
The main reason that process safety was a separate department was to prevent production and R&D from having vested interest in how test measurement results were interpreted and used or ignored. If calorimetric data suggests that a particular process reaction can run away or if a reaction should be initiated and run at a lower temperature, managers personally responsible for productivity may object owing to increased plant time or lower processing yields. This is especially problematic if prior experience has never shown a hint of a hazard, yet. Or, incidents in the past were not taken seriously or properly understood. The phrase “we’ve always done it this way” can be a very difficult barrier to overcome. And even if overcome, can revert back to the old practices over time.
This forces management to deal with safety margins and acceptable risk. They should automatically understand that zero risk is not possible. However, they may look back over the production history and not realize that they spent too much time near the edge of disaster.
Unknown risks
Imagine wearing a blindfold while standing 2 meters from the rim of the Grand Canyon. Someone turns you around a few times to scramble your senses. Now, even while not knowing the location of the rim, it is possible to walk around blindfolded and not go over the edge. You could do this for a short or a long time period and not fall in. Slowly you begin to doubt the hazard is real since you have not gone over the edge. Soon the risk is forgotten in the frenzy to reduce costs. Then one day you fall into the canyon and on the way down you muse about your own folly.
US President Donald J, Trump has declared that the illicit synthetic opioid Fentanyl has been declared a weapon of mass destruction, WMD. Fentanyl precursors have been added to the Convention against Illicit Traffic in Narcotic Drugs and Psychotropic Substances of 1988. This WMD status is from casting about for justifications to be at war with Venezuela or other countries.
When you are in control of the world’s most powerful military, there must be tremendous temptation to use it to clobber someone. When you’re a hammer, everything looks like a nail.
Intermediates added to the Convention against Illicit Traffic in Narcotic Drugs and Psychotropic Substances of 1988:
“In 2017, two main precursors namely N-Phenethyl-4-piperidone (NPP) and 4-anilino-N-phenethylpiperidine (ANPP) were placed under international control. Since that time, traffickers have adapted their approach to use alternative precursor chemicals for fentanyl manufacture. Three of these precursor chemicals, norfentanyl, 4-AP and 1-boc-4-AP, have now been placed under international control.
The WMD designation is clearly about legally mobilizing the military to interdict transport of fentanyl into the USA. The choice of fentanyl as a drug to manufacture by the cartels comes down to the extremely high potency and the relative ease of manufacture. The high potency, 2 milligrams for a lethal dose, means that it can be highly diluted with another drug and add to the overall potency. The high potency also means that a great many doses can be transported in small containers that may be easier to disguise and transport.
The obvious downside to fentanyl distribution is for the user. How careful are the people who spike intermediate quantities of substances, e.g., heroin or coke, so as not to provide a toxic product when repackaged for individual doses? Fentanyl should be redesignated as a highly potent toxic substance outside of the health care industry. As a drug distributor you probably don’t want your customers falling over dead from your product. That’s bad for repeat business. Regardless, user safety is unlikely to be a major concern to the distribution chain.
Sodium borohydride (NaBH4) can be used in the synthesis of fentanyl, so it is on the DEA Special Surveillance List. Sodium borohydride is a very useful and relatively safe hydride reducing agent that I and hundreds of thousands of others have used over the years in chemical synthesis. Sorry to see possible restrictions on its use.
The problem with this “designation of fentanyl and precursors” is that making a designer drug not cited in some list based the structure of fentanyl is that potent analogs can be dreamt up and produced if the right raw materials are available. Any organic or medicinal chemist should be able to come up with a list of candidates. Using existing drugs as a rough guide, producing obscure analogs is a skill set used by pharma companies frequently: Methyl, ethyl, butyl, futile … as the saying goes.
Wouldn’t it be nice to make some headway on the demand side too?
Fentanyl anecdote-
A few months ago I had surgery that involved my being anesthetized with fentanyl. I’ve been dosed with fentanyl several times and can report that it works well. What I didn’t note until the last instance was that it caused my face to itch badly for about 1 hour after surgery. Turns out this is a normal side effect and is not harmful. I had to wonder if addicts whose heroin was spiked with fentanyl had to suffer from both opioid-caused constipation and an itchy face. They have my sympathies there.
On CNBC today there was a report describing a looming shortage of trinitrotoluene, TNT, in the USA. According to this report, the USA quit manufacturing TNT in favor of importing it around 1986. Weapons-related consumption of TNT occurs both in military explosives for US stockpiles and for exported munitions. With Putin’s ridiculous war against Ukraine, America’s export of TNT-related munitions has increased, depleting the national inventory.
According to CNBC, the USA has been importing TNT from countries like Poland, Turkey, South Korea, Australia, and India. Recently, the price of TNT has increased to $20 per pound, increasing the cost of blowing rock, tanks and people to smithereens.
Some business considerations
If the competitive price for TNT is $20 per pound, then you want to ship it at a cost of at least $10 per pound. Even better would be $5-$8 per pound. This would be wonderful, but the fact is that the commodity chemical business is a high volume, low margin business. A margin squeeze is to be expected. Margins of a few dollars per pound wouldn’t be unusual for commodity chemicals.
Product below specifications can either be reworked or sold as a lower grade of product if there is demand. Commonly, below spec product can be blended with above spec product to pass QA. A TNT production plant coming online will increase the amount of product in the market, leading to depressed prices. What I’ve just said applies to the chemical industry at large, not just for explosives like TNT.
Unlike products such as bulldozers and trucks that leave the plant and go to work to create wealth, military armaments are not tools for wealth creation when used. They are consumables made for offensive or defensive destruction. Mining explosives are used to create wealth, but artillery shells are spent in conflict.
In the meantime, in Kentucky
In response to the precarious dependence on foreign vendors, the US government has awarded a contract to Repkon USA to construct a TNT manufacturing facility in Graham, KY. None other than the elderly Kentucky Senator Mitch “Grandpa” McConnell was present for the ceremony in Kentucky.
A bit of nitroaromatic history
TNT was first synthesized in 1863 by German chemist Julius Wilbrand during research in the area of synthetic yellow dyes. It wasn’t until 1891 that the explosive properties of TNT were discovered by German chemist Carl Häussermann. The earliest reported use of TNT as a military explosive was in 1902 and was used to fill artillery shells. As luck would have it, TNT is relatively insensitive and can be safely melted and poured into artillery shells or other munitions. According to Wikipedia, unlike the British explosive Lyddite, aka picric acid, TNT-filled artillery shells would not explode in contact with ships. Rather, TNT could withstand penetration of armor and then detonate internally. Artillery shells filled with the more sensitive picric acid would explode on contact with armor and explode externally, wasting its energy.
The older cousin of TNT, Picric acid, was used in the Battle of Omdurman, the Second Boer War, the Russo-Japanese War, and World War I. Picric acid was first synthesized from indigo by Peter Woulfe in 1771. It was synthesized purposely in 1841 by French chemist Jean-Baptiste Dumas. Of interest is the fact that the synthesis of indigo and other dyes was a target of much experimentation in Germany in the 1800’s.
Nitroglycerin
A quote from Wikipedia “The nitration of glycerin in 1846 by Ascanio Sobrero. He initially called it ‘pyroglycerine‘, and warned vigorously against its use. In fact, he was so frightened by what he created that he kept it a secret for over a year” (Wikipedia). Nitroglycerin is a nitrate ester wherein the three carbon atoms of glycerin are connected to the nitrogen through an oxygen atom. while TNT is a nitro compound with the nitrogen is connected to carbon atoms of toluene. The great sensitivity of nitroglycerin lies in the C-O-N connections while TNT and picric acid have C-N connections. Nitroglycerin is classified as a “nitro ester” while TNT and picric acid are “nitro aromatics.” The nitro ester functionality is much more susceptible to rapid disassembly by a stimulus like mechanical shock or heat.
The nitration of aromatic substances like benzene, phenol and toluene led to the introduction of powerful and relatively easy to manufacture explosives. Naturally, substances that are explosive attract great attention and have undergone a high degree of practical use to perfect.
People routinely disregard the ills of society, but when it comes to developing weapons of war, we become freaking Leonardo Di Vinci.
WWI saw the wide use of picric-acid-filled artillery shells that produced a new degree of violent destruction where used. At the same time the gas automatic machine gun, invented by Hiram Maxim, was introduced into warfare. WWI set the standard for violent death with the introduction of the Maxim machine gun and high explosives.
Interestingly, there is a medical use for nitroglycerin. It is used to treat angina. I have some at home myself, though I’ve never had to use it, thankfully.
Some words about nitration
Nitration of alcohols and aromatic compounds requires a source of -NO2, usually it’s nitric acid. However, the nitrating ability of nitric acid alone is weak, rather it must be activated to produce a more reactive form of nitrate. Sulfuric acid is a stronger acid than nitric acid and consequently is able to remove an oxygen atom by dehydration of the O-H group of nitrate anion producing water and affording a highly reactive (NO2)+ cation. The 6 electrons sandwiching the carbon skeleton of aromatic rings as with TNT, etc., are susceptible to attack by positively charged species and the (NO2)+ cation does the job. The advantage of nitric acid in all practicality is that a hydrogen atom, H+, is already attached producing a water molecule that will easily detach from the nitrogen to form the reactive species, (NO2)+. [As an aside, in chemical processing, liquids are easier and safer to transfer by pumps and piping as opposed to solid addition.]
Formation of nitro esters and nitro aromatics. Graphics by Buford Pusser.
The production of TNT might seem fairly simple- all that is needed are the cheap commodity chemicals toluene, sulfuric acid and nitric acid plus reactors and other process equipment that can resist strongly corrosive acids. The scale of the process will need to be large enough to capture the economies of scale in accordance with capital costs. Utilities like heating and chilling will be needed as well as possible on-site water treatment if allowed. And don’t forget an idiot-proof written procedure and EHS staff as well as talented management. A properly equipped analytical lab will be required for QA/QC.
More fundamentally you’ll need a remote site on which to build a plant that is supplied with sufficient electrical power as well as water and sewer. The state, county and nearby towns will insist on iron-clad assurances of worker safety and proper hazardous waste management. The state will be watching air emissions closely. Then there is finding an insurer to cover the plant and operations.
If you start a nitration operation, why not plan for products in addition to TNT? It can be unwise to operate a 1 act pony. What if the pony dies?
Today I have a slightly different demographic of readers of this blog than in the past, so I’ve been dredging up old posts into the light of day. This is a renamed post from September 3, 2011. I’ve changed some wording to be a bit more mellifluous if that’s even possible.
==========
I’ve had this notion (a conceit, really) that as someone from both academia and industry, I should reach out to my colleagues in academia in order to bring some awareness of how chemistry is conducted off-campus. After many, many conversations, an accumulating pile of work in local ACS section activities, and visits to schools, what I’ve found is not what I expected. I expected a bit more academic curiosity about how large-scale chemical manufacturing and commerce works and perhaps what life is like at a chemical plant. I’d guessed that my academic associates might be intrigued by the marvels of the global chemical manufacturing complex and product process development. Many academics would rather not get all grubby with filthy lucre. Not surprisingly, though, they already have enough to stay on top of.
What I’ve found is more along the lines of polite disinterest. I’ve sensed this all along, but I’d been trying to sustain the hope that if only I could use the right words, I might elicit some interest in how manufacturing works- that I could strike some kind of spark. But what I’ve found is just how insular the magisterium of academia really is. The walls of the fortress are very thick. I’m on a reductionist jsg right now so I’ll declare that chemistry curricula is firmly in place on the three pillars of chemistry- theory, synthesis, and analysis. In truth, textbooks often set the structure of courses. A four-year ACS certified chemistry curriculum spares only a tiny bit of room for applied science. I certainly cannot begrudge departments for structuring around that format. Professors who can include much outside the usual range of academic chemistry seem scarce.
It could easily be argued that the other magisteria of industry and government are the same way. Well, except for one niggling detail. Academia supplies educated people to the other great domains comprising society. We seem to be left with the standard academic image of what a chemical scientist should look like going deeply into the next 50 years. Professors are scholars and they produce what they best understand- more scholars in their own image. This is only natural. I’ve done a bit of it myself.
Here is my sweeping claim (imagine waving hands overhead)- on a number’s basis, chemists apparently aren’t that aware of industrial chemical synthesis as they come out of a BA/BS program. That is my conclusion based on interviewing many fresh chemistry graduates. I’ve interviewed BA/BS chemists who have had undergraduate research experience in nanomaterials and atomic force microscopy but could not draw a reaction scheme for the Fisher esterification to form ethyl acetate, much less identify the peaks on 1HNMR. As a former organic assistant prof, I find it sobering and a little unexpected.
A mechanistic understanding of carbon chemistry is one of the keepsakes of a year of sophomore organic chemistry. It is a window into the Ångstrom-scale machinations of nature. The good news is that the forgetful job candidate usually can be coached into remembering the chemistry. After a year of sophomore Orgo, most students are just glad the ordeal is over and they still may not be out of the running for medical school.
I think the apparent lack of interest in industry is because few have even the slightest idea of what is done in a chemical plant and how chemists are woven into operations.
To a large extent, the chemical industry is concerned with making stuff. So perhaps it is only natural that most academic chemists (in my limited sample set) aren’t that keen on anything greater than a superficial view of the manufacturing world. I understand this and acknowledge reality. But it is a shame that institutional inertia is so large in magnitude in this. Chemical industry needs chemists of all sorts who are willing to help rebuild and sustain manufacturing in North America. We need startups with cutting edge technology, but we also need companies who are able to produce the fine chemical items of commerce. Have you tried to find a company willing and able to do bromination in the USA lately? A great deal of small molecule manufacture has moved offshore.
Offshoring of chemical manufacturing was not led by chemists. It was conceived of by spreadsheeting MBAs, C-suite engineers and boards of directors. It has been a cost saving measure that mathematically made sense on spreadsheets and PowerPoint slide decks. The capital costs of expansion of capacity could be borne by others in exchange for supply contracts. There is nothing mathematically wrong with this idea. Afterall, corporate officers have a fiduciary responsibility to their shareholders. Allowing profit opportunities to pass by is not the way to climb the corporate ladder.
We have become dependent on foreign suppliers in key areas who have control over our raw material supply. Part of control is having manufacturing capacity and closer access to basic feedstocks.
The gap between academia and industry is mainly cultural. But it is a big gap that may not be surmountable, and I’m not sure that the parties want to mix. But, I’ll keep trying.
[Note: This post is about replacing the hydrogen atoms along the carbon backbone of a polyolefin polymer with fluorine atoms to produce a fluorocarbon surface on a finished good. Here “finished good” refers to anything from polyolefin pellets, powders, components or blow molded articles such as HDPE bottles.]
Recent news has highlighted the use of fluorinated High-Density Polyethylene (HDPE) packaging for pesticides and other products, bringing more attention to the issue of PFAS/PFOS contamination.
What we’re not talking about is a polymer made from fluorinated monomers or comonomers. This refers to a hydrocarbon HDPE bottle made from ethylene (H2C=CH2) monomer that is fluorinated after the bottle is manufactured.
What’s more, the HDPE fluorination process is said to produce PFAS/PFOS (how?) substances that can migrate. Although this technology is not new, and fluorinated hydrocarbon bottles have been around well before the widespread concern over PFAS/PFOS residues, the significance of such contamination was not fully anticipated. As a chemist, the extensive release of fluorinated low molecular weight alkyl derivatives like PFAS/PFOS came as a surprise to me despite knowing that an analogous situation with fluorinated pharmaceuticals that are getting through wastewater plants due to their resistance to microbiological decomposition. For myself only, very little concern for PFAS/PFOS pollution has been noted. You might suppose that chemists could have led the way to understanding. But, not to my knowledge.
The perfluorinated alkyl materials in question bear a close resemblance to TeflonTM which is known for its chemical inertness and lubricity. In chemistry, Teflon is usually ignored as unreactive with most chemicals, except perhaps molten alkali metals. Strategically placed fluorinated features on a molecule can lend the property of greater hydrophobicity or lipophobicity with increased electron withdrawing properties. The high electronegativity of fluorine pulls electron density towards the fluorine atoms through the sigma bonds of a molecular skeleton. Fluorinated organic acids very often have dramatically increased acidity like triflic acid, CF3SO3H, or increased alkylating reactivity like magic methyl, F-SO2(OCH3). By contrast, fluorinated carbon chains themselves are fairly unreactive and quite hydrophobic, as in water repellant. The water repellency of fluorinated hydrocarbons is a very attractive property commercially.
Below are images of the hydrocarbon hexane in ball and stick form and below in a space filling rendering. To the right is perfluorohexane and below that is its space filling rendering. Hexane is just an example of an “ordinary” hydrocarbon that could be perfluorinated.
Graphics by Sam Hill. Hexane (left) and perfluorohexane (right). As can be seen on the right, the green fluorine atoms are rendered larger than the corresponding white hydrogen atoms because fluorine atoms are larger than hydrogen atoms. In some rendering software, the space filling structures are adjusted to show where some percentage (i.e., 95 %) of the electron density is located. These renderings are by ChemSketch so God only knows how atoms are scaled.
A brief interlude on molecular polarity
Before we go on, there is the matter of polarity, dipolarity, dipolar chemical bonds and dipolar molecules. A dipolar polar chemical bond is one in which the distribution of electrons is lop-sided. That is, one atom of a chemical bond has a bit more negative charge than the other, which is thereby deficient in negative charge, or by default carrying a partial positive charge. Chemical bonds, functional groups and entire molecules can be dipolar.
But charge comes in whole numbers, so how can we talk about partial charge? A covalent chemical bond consisting of 2 atoms, same or different, will hold together because the two atoms share a pair of outer electrons. If one of the two atoms in the bond has a greater affinity for negative charge, then the cloud of 2 bonding electrons will spend a bit more time near the more electronegative atom. This shift leaves the other nucleus slightly deficient of negative charge averaged over time meaning that the positive charge of the nucleus is slightly more exposed to the world.
Graphics by Jed Klampett. Polar and nonpolar molecules.
In chemistry there is a saying- “likes dissolve likes”. This means that a polar solvent like water can more readily dissolve polar solids and may mix freely with other polar liquids. Nonpolar liquids like hydrocarbons can dissolve nonpolar solids and may mix freely with other nonpolar liquids. Amphiphilic substances have both polar and non-polar features allowing them to compatibilize polar and nonpolar molecules together. Soaps and detergents are in this category.
We should be careful here. The polar-polar and nonpolar-nonpolar solubility generalizations above are really just bookends across a vast open shelf of partial solubilities between them. Nonetheless, it is a useful rule of thumb.
So, if likes dissolve likes, and the fluorine atoms on a molecule accumulate a bit of negative charge, then why doesn’t a fully fluorinated organic molecule freely dissolve in water owing to fluorine’s negative polarity via hydrogen bonding with water’s positively polarized hydrogen atoms?
Carbon atoms can form bonds with itself or other atoms in several ways that give rise to different overall shapes.
Back to our regularly scheduled content
In situ fluorinated packaging, a niche within the packaging industry, was not something I was fully cognizant of until recently. I have come to understand that HDPE, along with numerous other polymers, can undergo treatment with elemental fluorine or fluorinated reagents to alter the hydrocarbon polymer’s C-H groups and convert them into C-F groups. This alteration gives the HDPE surface properties similar to a perfluorocarbon like Teflon™. For HDPE pesticide packaging, this fluorocarbon layer reduces the product’s permeability to the pesticide’s components. Package fluorination is all about reducing permeability of the container.
Definition: Hydrocarbon. A category of substances composed only of hydrogen (H) and carbon (C). There are 4 main sub-categories: Alkanes, alkenes, alkynes, and aromatics. A hydrocarbon can be composed of any combination of the 4. The principal mineral sources of hydrocarbons are coal, petroleum and natural gas. A hydrocarbon may also be called an organic substance, where organic refers to a carbon-based substance.
HDPE, high density polyethylene, is a hydrocarbon polymer of ethylene gas and often with various hydrocarbon comonomers. Hydrocarbon polymers, also called polyolefins, are notable for their considerably inert chemical properties. Inertness is the resistance to chemical change. However, contact with certain fluorinating agents like F2, ClF3, NF3, etc., diluted in an inert gas can, at relatively low temperatures, exchange the H atoms of HDPE with F atoms. Eventually, all or most of the H atoms on the polymer surface will be exchanged. A carbon molecule that has F atoms replacing all H atoms is said to be perfluorinated.
Pesticides are meant to be spread over selected parts of the environment to do their trick. A great many pesticides are synthetic organic chemicals so naturally there is the possibility of any given pesticide or solvent to diffuse through a hydrocarbon-based container. Migration of product molecules into the polyolefin packaging, in this case (HDPE), can result in the release of the hazardous contents and compromise the overall containment, possibly resulting in exposure to the public and the environment.
It should be possible to slow the rate of diffusion of any given hazardous material through a non-fluorinated container by simply making the container walls thicker. The polyolefin manufacturers would be in favor of this, but the converters who buy the plastic pellets to blow mold the containers may balk. Their raw material costs would rise and they would have to pass the costs to customers, who will resist the cost increase. Then with the increase in mass flow of polymer melt necessary, perhaps the throughput or required extruder torque might change unfavorably. Hard to say.
Some of the small-molecule bad actors
On March 5, 2021, EPA published the list below of PFAS/PFOS compounds found in the 20-50 ppb level in fluorinated HDPE containers used to store and transport a mosquito control pesticide product.
Abbreviated
Full Name
PFBA
Perfluoro-butanoic acid
PFPeA
Perfluoro-pentanoic acid
PFHxA
Perfluoro-hexanoic acid
PFHpA
Perfluoro-heptanoic acid
PFOA
Perfluoro-octanoic acid
PFNA
Perfluoro-nananoic acid
PFDA
Perfluoro-decanoic acid
PFUdA
Perfluoro-undecanoic acid
These are all perfluoroalkyl carboxylic acids listed by increasing chain length. Notably the terminal carbon is fully oxidized to the carboxylic acid and is not fluorinated. This acidic end gives a chemically reactive handle for further manipulation of the PFAS/PFOS if desired.
PFOA, perfluorooctanoic acid, has been industrially produced by what is now 3M since the mid-1940s. It has been used to place TeflonTM coatings on frying pans. It was originally prepared by the electrochemical fluorination (ECF) of octanoyl (ock TAN oh ill) chloride, the hydrogen saturated 8-carbon acid chloride. ECF produces the perfluorooctanoyl fluoride which is then hydrolyzed to the acid chloride liberating HF.
Perfluorination of HDPE bottles relies on the most electronegative element, diatomic fluorine gas, F2, or other similarly reactive fluorinating reagents, and does chemistry on a solid polyolefin surface. Fluorine gas is diluted in a suitably noninterfering gas like nitrogen, argon or CO2 and then exposed to the polymer of interest at a prescribed pressure, temperature and exposure time. Fluorine atoms replace hydrogen atoms on the polymer chain. According to one source, the rate of fluorination is diffusion limited. This means that the fluorination reaction is very fast. The presence of molecular oxygen with molecular fluorine had a retarding effect on fluorination proportional to the concentration of oxygen gas. The presence of oxygen led to it being incorporated onto the polymer.
Given the advantage of impermeability provided by fluorinated polyolefin articles, it is clear that there are many excellent applications of in situ fluorinated polyolefins. The replacement of glass and metal with lighter fluorinated HDPE containers may save on transportation costs on a weight basis. Whether or not the economics favor fluorinated polyolefins over glass or metal manufacturing costs kg for kg is unclear.
The range of application categories listed above is quite large. Each entry in the list has many individual components that may be subject to fluorination as well. It is no wonder that PFAS contaminants are spread widely around the world. The US EPA has issued a letter (below) to companies fluorinating HDPE to beware of accidentally producing PFAS/PFOS in their operations. Specifically warning about the connection of PFAS formation caused by the inclusion of oxygen in the fluorination process. The letter specifically cites “EPA’s 2020 long-chain perfluoroalkyl carboxylate (LCPFAC) Significant New Use Rule (SNUR) (40CFR § 721.10536), that are found to be present in or on fluorinated polyolefins may be subject to TSCA regulations and enforcement.”
“It is during certain types of fluorination (e.g., the presence of oxygen) that the manufacture of PFAS has occurred. Manufacturers (including importers), processors, distributors, users, and those that dispose of fluorinated HDPE containers should be reminded of this potential for manufacturing PFAS and comply with any applicable regulations under TSCA, as described in the next section.“
“EPA is aware of alternative fluorination processes that use fluorine gas in the presence of gaseous inerting (e.g., nitrogen) without the presence of oxygen that could reduce the potential for unintentional manufacture of PFAS. These alternative processes for fluorination of polyethylene are highlighted in the U.S. Food and Drug Administration’s (FDA) August 2021 letter on this issue as it relates to food contact articles.”
“Requirements under TSCA PFAS Significant New Use Rules. Certain PFAS, including long-chain PFAS as defined in EPA’s 2020 long-chain perfluoroalkyl carboxylate (LCPFAC) Significant New Use Rule (SNUR) (40 CFR § 721.10536), that are found to be present in or on fluorinated polyolefins may be subject to TSCA regulations and enforcement. EPA considers the manufacturing of certain PFAS from the fluorination of polyolefins to be a significant new use under TSCA. LCPFAC chemical substances present in polyolefins due to the fluorination process would be considered byproducts of the manufacturing process because they are produced during the manufacture of the fluorinated polyolefins and do not have a separate commercial intent (40 CFR § 720.3(d)). LCPFAC chemical substances that are byproducts of the manufacturing process for fluorinated polyolefins do not meet the requirements of the byproducts exemption at 40 CFR § 721.45(e)5 and are subject to significant new use notice requirements. Significant new use rules require industry to notify EPA at least 90 days before commencing the manufacturer including import or processing of subject chemical substances for a significant new use. The required significant new use notification (SNUN) initiates EPA’s evaluation of the conditions of use associated with the significant new use. Entities may not commence manufacturing (including import) or processing for the significant new use until EPA has conducted a review of the notice, made an appropriate determination on the notice, and taken such actions as are required in association with that determination. Tala R. Henry, Ph.D., Deputy Director Office of Pollution Prevention & Toxics 2022/03/24.”
Fluorination and fluoridation. What’s the difference?
So we do not make people worried about their fluoride toothpaste or their fluoridated drinking water, let’s sort this out. Toothpaste and drinking water have a soluble ionic fluoride salt like sodium fluoride, NaF, or sodium monofluorophosphate, sodium MFP or chemically Na2PO3F. Sodium MFP is water soluble but not stable in water. It hydrolyzes to release fluoride by displacement by water to form dibasic phosphate. The MFP hydrolysis reaction is: PO3F2− + HO– → HPO42− + F−. The fluoride anion, F–, is not nearly the same as fluorine gas, F2. The F– ion bumps into tooth enamel where it binds tightly with calcium in the tooth: Ca5(PO4)3+(aq) + F−(aq) → Ca5(PO4)3F(s). This is the context in which the word “fluoridation” is used. Fluoride ions bond tightly to calcium++ ions in general. Fluoridation is just a specialized variety of fluorination and is mostly confined to the area of water treatment and toothpaste.
Fluorination is a chemical process wherein fluorine atoms are added to chemical compounds. Contact between organic substances and pure elemental fluorine gas is extremely exothermic and sometimes explosive. The dilution of F2 gas with an inert gas like nitrogen, helium or argon has a thermal safety component as well.
Polymer fluorinationout in the world- Patents
One source of manufacturing information about proprietary articles and processes is the US Patent and Trademark Office, USPTO. In order to secure your legal right to a patent, the patent applicant must disclose the exact art that is being claimed. This is because the world must have a fair chance to avoid infringement. Google Patents provides the exact text of individual patents, US and others. It also provides a timeline showing the ownership of the patent and whether or not the patent is active, expired or abandoned. Google patents also provide links to patents cited in the patent and patents that have cited the instant example.
Being a Google product, Google Patents has extensive and flexible search capacity. Rather than attempt to make a list, it is a better use of the reader’s time to go to the site yourselves and explore. Note that a search will find patents from all over the world as well as patent applications. Google patent provides a English translated version of the patent.
In searching for patents claiming compositions and methods around the fluorination of polymers, more than a few patents can be found. One can search for patents using the USPTO website (obviously) or from Google Patents.
Another good place to look for relevant art is from a patent you have already pulled up in Google Patents. Near the bottom of the patent from Google Patents is a section labeled “Patent Citations.” This section list prior art patents disclosed by the assignee and those found by the patent examiner in the course of the examination process. Prior art is disclosed by the assignee in the granted patent as well, but in Google Patents there are hotlinks to patents to aid the convenience factor.
In situ fluorination
There are companies who will fluorinate the surface(s) of High-Density PolyEthylene (HDPE) and PolyPropylene (PP) containers. HDPE and PP are especially of interest owing to their utility in packaging liquids. These two polymer classes have great rigidity and strength and are in wide use. However, they share certain weaknesses such as air permeability and permeability of the contents. Air permeability is highly undesired in food packaging as it allows for reduced shelf life or customer satisfaction with the contents. Food and drugs may be susceptible to air degradation and possible reduction of shelf life.
Note: The expiration date on a product does not necessarily mean that the product will go bad when that date arrives. The day after the expiration date is that date at which the manufacturer/seller will no longer guarantee “freshness” or some other type of quality. For instance, normal pasteurized milk is not sterile. Pasteurized milk should be good up to 1 week past the code date as long as it has not been allowed to warm up or been contaminated. Once the milk has warmed to room temperature, the normal bacteria loading will enter log-phase growth and could spoil within 1 day.
In situ fluorination is process wherein hydrocarbon polymer containers are exposed to diluted fluorine gas at a specified temperature for a specified time. At the surface hydrogen atoms along the length of the polymer are replaced with fluorine atoms. The result is a polymer along the surface which resembles TeflonTM to some extent. Some of the desirable properties of TeflonTM are then taken on by the HDPE or PP surface. This H/F exchange at the surface does not affect the properties of the base polymer.
There is one caveat, however. The fluorination must be performed with the exclusion of oxygen. One source says that the vacuum chamber in which the fluorination will take place must be pumped down to 0.1 Torr of residual air prior to exposure to fluorene gas.
Fluorination patents
Below us from the description in US5274049A Filing date 1991-07-19, Application filed by SHAMBAN WILLIAM S, W S SHAMBAN AND Co.
A method for the direct fluorination of elastomers “in order to reduce the static and dynamic friction characteristics and to increase the wear life and abrasion resistance of the elastomers. The invention also relates to elastomeric articles modified by the fluorination method.”
“What is claimed is:
1. A method of producing fluorinated elastomeric articles, consisting essentially of the following steps:
providing an elastomeric article, said elastomeric article comprising an elastomeric polymer having a backbone chain having a plurality of hydrogen atoms attached thereto;and
exposing said elastomeric article to gaseous fluorine under conditions sufficient to reduce the friction coefficient of said article without promoting degradation of the tensile properties of said article.”
Claim 8 claims a method using a hydrogen fluoride scavenger …
“8. A method for producing a fluorinated elastomeric article having a reduced coefficient of friction, comprising the steps of:
placing a thermoset elastomeric article and a hydrogen fluoride scavenger in a closed reactor vessel, said thermoset elastomeric article comprising an elastomeric base polymer having a backbone chain, said backbone chain including sufficient carbon atoms having replaceable aliphatic carbon-hydrogen bonds so that a fluorinated matrix of said fluorinated elastomeric article reduces said coefficient of friction;”
In the description the patent cites sodium fluoride, NaF, as an HF scavenger wherein NaF + HF => Na[HF2], sodium bifluoride.
Inhance Technologies LLC filed application US20190040219A1, but it was later it was abandoned due to failure to respond to an office action. The application claimed a multistep method for fluorinating elastomeric workpieces with 20 % F2 in nitrogen and “altering certain mechanical properties such as tensile property [and] the elastic modulus, an impact property, a wear property, etc.“
Systems and methods for processing fluoropolymer materials and related workpieces, US11879025, filed 2021-04-23, Current Assignee: Inhance Technologies LLC. Claims method of removing perfluorinated compounds from fluoropolymers. The core of the art involves placing a fluoropolymer work piece in a thoroughly deoxygenated chamber, heated from 25 C to 300 C and exposed to a fluorinating atmosphere such as F2/N2 for specified time period. This treatment is claimed to remove fluorocarbons like PFOA to non-detectable levels. There is no mention of where the PFOA goes afterwards, but it looks promising if accurate. However, the granted patent is off-limits for 20 years unless a license is obtained or some other arrangement is made.
Fluorination is imbedded deeply into the design of a great many articles of commerce. The water repellency of perfluorinated polymers in fabrics is one of the chief applications of fluorinated organic materials. The inherent lubricity of PTFE, its built-in chemical inertness and its hydrophobicity have ingratiated millions of consumers and have met performance expectations world wide.
Perfluorinated foams for fire protection in aircraft hangers and industrial spaces are valuable for their ability to float on the surface of burning liquid fuels, blanketing the surface as a vapor and oxygen barrier. The suppression of flammable volatiles in a fire by a layer of protective foam can inhibit flashover of the fire, reducing the overall damage of a fire. The fire retardancy of perfluorinated substances inhibits their combustion and discourages continued burning when the flame source is removed. Halogens as a group have been used for fire retardancy and with bromine in particular.
The chemical origin of the fire retardancy properties of perfluorinated organic materials lies in the low reactivity of the -CF2– fluorine atoms with oxygen. In the combustion of hydrocarbons, hydrogen atoms are readily removed by oxygen or radical species to form water. The C-F bond is one of the strongest bonds in organic chemistry and is slow to be removed by oxygen.
Drug molecules are frequently fluorinated in particular locations on the drug molecule. A C-F bond resists catabolic degradation and enhances the local hydrophobicity of the drug allowing for greater half-life and enhanced drug potency. The down side is the resistance to catabolic degradation and excretion. Many drug molecules are released intact into sewage treatment facilities where they also resist degradation, possibly due in part to the fluorinated features. The effect is that fish and other organisms are exposed to the drug. As with humans, fish and other creatures of the waterways and soil did not evolve with biochemical mechanisms to deal with fluorinated organics.
In the in situ fluorination process, PFAS/PFOS side products can form, especially when oxygen is present. This can be monitored by quality control but companies will comply with recommended PFAS/PFOS best practices only if there are regulations or the threat of them. Nations regulating PFAS/PFOS contamination will have to compete with nations who do not impose regulations. This is the usual scenario for nations with heavy reliance on imported articles but uneven regulation.
The state of Nevada is quickly becoming the leading source of lithium in the USA and beyond. In the state there will soon be three major types of lithium ore beneficiation- Brine evaporation, hard rock extraction and lithium clay extraction. Nevada already has in excess of 180,000 active mining claims amounting to 49 % of the total BLM national inventory. In addition to this, Nevada has “198 authorized mining plans of operations, and 282 active exploration notices.” Nevada has a long history of fruitful gold and silver mining.
Nevada had earlier won the gold deposit lottery with the Carlin Trend occupying much of the northwestern section of the state. The Carlin Trend has become an archetype in gold mining. These deposits are often described as Carlin-type “invisible gold” ore deposits. Such a deposit is characterized as sediment hosted and disseminated [Editor: disseminated seems like a bummer]. Gold in such deposits are typically invisible and often only detected by lab analysis. According to Wikipedia, most of the gold mines in the Great Basin of the western US are of the Carlin-type.
But, enough about gold and on to lithium
After 6 years of regulatory scrutiny, a new lithium-boron open-pit mining operation in Nevada operated by Australian mining company ioneer has just been approved by the Bureau of Land Management, BLM, for Rhyolite Ridge. The mine is located in the Basin and Range Province near the southwest border of Nevada and California.
The old joke used to be that a mine is a hole in the ground with a liar standing at the top. With the independent economic evaluations available today as part of the disclosure to investors, the likelihood of being duped by fake or salted deposits has dropped considerably. However, the market value of the ore in the future is still subject rapid and unpredictable change.
If you find yourself flying over Nevada on a clear day, you can easily see the basin and range features of the terrain. Nevada occupies only a small part of the total area. The basin and range province extends north to the Columbia Plateau and south into the Central Mexican Plateau.
A very small part of Nevada’s basin and range landscape as viewed from above the Rhyolite Ridge area in Nevada. Image from Google Maps.
The Basin and Range Province of North America. Image from Wikipedia.
Rhyolite Ridge Lithium-Boron Project
The BLM approval opened up $1.19 billion of potential funding of which $700 million is from a US government loan. According to Mining.com, Rhyolite Ridge is the first new lithium mine in 60 years and the first new boron mine in the last century in the US. [Note: I have to assume “new” means new hard rock mineas opposed to brines or evaporites] While the approval by BLM has opened some doors to funds, not everyone is convinced of the major investor’s liquidity.
So, what is rhyolite?
I can’t improve on the definition found in Wikipedia, so I’ll just quote it with the links intact-
If you have ever seen molten glass and noticed its high viscosity, this gives an idea of what high silica content does to lava. The higher viscosity provided by the silica component suppresses the release of gases until nearer the surface where they are released as bubbles with vigor. It is very much like a comparison between boiling pasta water and boiling marinara sauce. The marinara sauce spatters badly due to its viscosity but the pasta water just does a rolling boil.
Source: Mashed.com. Spattering is a universal behavior of hot, gassy fluids. In this case the gas is steam. Magma also contains steam.
The Rhyolite Ridge lithium-boron (LiB) deposit is said by some to be the only known LiB deposit in the US and only one of two known in the world.
“The Rhyolite Ridge mine will run for 22 years. It will produce 22,000 tonnes of lithium carbonate a year. That’s enough to power 370,000 electric vehicles. It will also produce 170,000 tonnes of boric acid, according to the company. The boron contributes 30% to 40% of the mine’s revenue, providing a buffer against lithium market volatility.” –Mining.com
Around the world new economic lithium deposits are being discovered now and then, and a few are being readied for mining. It was announced recently that BLM has approved operations at the Rhyolite Ridge Lithium-Boron Project in southwestern Nevada.
What is interesting about this Rhyolite Ridge project is that it aims to produce both lithium and boron. I’m not an engineer so maybe I’m overly impressed, but the processing plant they propose seems very clever. They will produce their own sulfuric acid from sulfur and extract waste heat for use in generating steam for evaporation of the extracts and electricity. They are completely off the energy grid.
The extracted ore, now called ROM or run-of-mine, is transported to the plant straight from the mine and sized by crushing to 20 mm pieces. The crushed ROM is then taken to a series of sulfuric acid extraction vats and leached for ~ 7 days. The pregnant leach solution containing the lithium, boron and soluble impurities is then taken to evaporators with repeated crystallizations and, using differential solubility, separates the lithium component from the boric acid. In the end they produce lithium carbonate. The video does show a soda ash (sodium carbonate), Na2CO3, silo so I assume that is where the carbonate comes from to produce lithium carbonate, Li2CO3 and to neutralize residual sulfuric acid.
Silver Peak Lithium Brine
Of interest is the nearby Silver Peak lithium brine operation operated by Albemarle just a few miles to the north of Rhyolite Ridge. The Google Maps image below shows the evaporation ponds at the Silver Peak lithium operation. Silver Peak produces both technical grade lithium carbonate and lithium hydroxide.
Image from the Operational Land Imager-2 (OLI-2) on NASAs Landsat 9. A view from space of the Silver Peak lithium brine evaporation ponds in SW Nevada.
McDermitt Caldera: Thacker pass
Another large lithium deposit was discovered in the McDermitt Caldera along the Nevada-Oregon border. Within the caldera is the Thacker Pass Lithium Mine. This lithium deposit was approved for open-pit mining by BLM on January 15, 2021, though it has been plagued by protests and an injunction. As with the rest of the McDermitt Caldera lithium, the Thacker Pass lithium is described as a lithium rich clay deposit. This is unique for lithium mines since brine extraction and hard rock mining of spodumene have been the norm. Thacker Pass’ lithium deposit is the largest known volcano sedimentary deposit in the US at an average grade of 0.22 %.
In 2023 GM invested $650 million in the Canadian Lithium Americas Corp. The Thacker pass operation is through its wholly owned subsidiary Nevada Lithium, LLC, which is responsible for production. Car giant GM’s investment gives them exclusive access through the first phase of production. Lithium Americans has received a conditional approval for a $2.2 billion loan from the US Department of Energy.
The Thacker Pass measured and indicated lithium resources are 13.7 million tons of lithium carbonate equivalent. Lithium Americas calculates that the recoverable lithium is worth $3.9 billion.
Interestingly, the McDermitt Caldera is possibly the oldest of a sequence of calderas produced by the Yellowstone Hotspot. The McDermitt Caldera amounts to a lava dome that collapsed ~16.4 million years ago forming a large caldera within which several smaller calderas have formed and in which later filled with water forming a lake over the tuffaceous ash. Over time the lake produced sediments that were deposited on the floor of the lake. The source rock is rhyolite which is usually the case in the state.
The Yellowstone hotspot stays relatively constant while the crust moves over it, leaving a trail of calderas and a record of volcanic activity on the surface behind. The McDermitt caldera is labeled ’16’. Source: National Park Service.
Briefly, the lithium clay is excavated, gravel and rocks are removed, and the clay is suspended in water to form a slurry. The slurry is leached by adding sulfuric acid that is produced on-site and the lithium in the ore is extracted into the acidic liquor. Finally, the dissolved lithium is recovered as lithium carbonate and lithium hydroxide. Gangue material is deposited back into the excavated sections of the mine.
The McDermitt caldera contains many breccia and fracture zones and associated with these are deposits of other metal ores. Specifically, mercury from cinnabar ore bodies and uranium from autunite (uranyl, U6+) (Ca(UO2)2(PO4)2·10–12H2O) and pitchblende (uranate, UO2, U4+) ore bodies. Mercury reserves in the caldera, according to USGS, are estimated to be 400,000 flasks.
A Mercurial Rambling
Mercury has been packaged (and still is) at 76 pounds to the flask. This measure got its name from the mining and smelting of cinnabar in the mountains of Peru. One site was particularly rich- Huancavelica, Peru. The Spaniards had known prior to their arrival to the New World that liquid mercury could dissolve gold from ore to produce what we now know as a mercury amalgam. With very strong heating the mercury could be driven off as vapor to recover the precious metal.
Spain had its own cinnabar mine in what is now Almadén, Spain, which produced over 250,000 tonnes of mercury. The Spaniards had considerable experience with mining, refining and using mercury prior to discovery of it in the new world. Mercury was quite valuable to the Spaniards but they were faced with transporting it from, say, Huancavelica, Peru, to the Carribean coast where their ships could load the mercury and distribute elsewhere. Anecdotally, it has been estimated that for every ounce of gold produced in the new world, ten ounces of mercury were consumed.
Luckily for the 49er gold rush miners, cinnabar had previously been discovered in California. You can actually visit the old New Almaden mine museum south of the Bay Area. It is worth a trip if you are in the area.
The Spaniards figured that a man could carry what turned out to be as much as 76 lbs of mercury through difficult terrain to the Caribbean coast. Even better for the Spaniards, they knew how much gold could be extracted per pound (or whatever unit) of the mercury they dispensed. This gave them an idea of how much gold to expect and closer control of the mines. By controlling mercury, they controlled who could use it for mining and how much gold they could recover.
Back to lithium
As of this writing, Albemarle is the largest producer of lithium in the US. But the largest known deposit in the US is at Thacker Pass. The Albemarle Silver Peak lithium brine operation is legally defined as placer mining whereas the Thacker Pass operation is lode mining. The word ‘legally’ is used because any claims filed are restricted to placer or lode mining- one does not transfer to the other.
A Word or Two About Rhyolite
Rhyolite is a type of volcanic rock characterized as a ‘fine-grained extrusive igneous rock‘. Its color can vary from pale light grey to pinkish when composed of mainly quartz and feldspar or dark when of a mafic, low silicate composition was present. A quiet eruption of lava gives a solid, denser rhyolite whereas in an explosive eruption can produce the vesicular pumice. The lower density of pumice allows it to float on water. Erupting volcanic islands can produce floating rafts of pumice in the nearby waters. As magma rises in the throat of a volcano, the pressure drops and dissolved gases can form bubbles which, if they fail to disengage from the magma completely, can be ejected into the cooler air and freeze into ‘foamy’ structure. [Note: the word ‘foamy’ is my own and earth science people cannot be blamedfor this.]
A Bit More About McDermitt Caldera
The United States Geological Survey (USGS) published open-file report 76-535 in 1976 titled Geology and Ore Deposits of the McDermitt Caldera, Nevada-Oregon. A 1978 USGS open-file report by Newmont Exploration, Ltd., 78-926, James J. Rytuba and Richard K. Glanzman, Relation of Mercury, Uranium and Lithium Deposits to the McDermitt Caldera Complex, Nevada-Oregon, goes into greater detail on the three minerals.
Summary: This essay addresses the important role the federal government has played in promoting the American march of progress. The old saying that “Necessity is the Mother of Invention” has a large element of truth to it. It is not enough to identify a problem or challenge. For a person, group or organization to solve a technological problem or challenge, the goal must be understood completely, resources acquired, a plan must be constructed and approved by those who control the purse strings, and skilled people must be organized and set to work on the matter at hand.
The federal government can provide the Necessity needed for attention and resources put to play in achieving a goal. For instance, NASA will set a goal and is able to open a project up for bid. The gov’t can provide seed money to the contractor for prototype equipment to present with their bid. Government grants provide the necessity to stimulate invention, hopefully on a competitive basis.
……………………
I have been a lifelong aerospace enthusiast from Project Mercury forward to the present day. What I’m realizing, however, is that I’m increasingly skeptical of the value of further manned space flight by NASA. Whatever the 6 successful manned Apollo landing missions on the moon may have found tramping around the regolith up there, evidently not enough value was found to compel the USA to go back. Obviously, the Apollo program was partially a geopolitical stunt to rival the USSR for prestige and by many measures the USA won. But what did we win? Prestige and a great many valuable technological spin-offs.
In the early 1960’s the US government financed and organized JFK’s challenge of landing a man on the moon and returning him safely to Earth by the end of the decade. Our government allocated considerable national treasure to the moon landing project and put lives on the line. Arguably, of greater importance than a round trip to the moon was the powerful boost to aerospace, computer, and other technologies. The technology-push advances funded by the government would soon become important economic drivers for industry.
In fulfilling Kennedy’s challenge there was both popular excitement about the space program and more than a little skepticism. The USA was increasingly bogged down with the Viet Nam war. Into the early 1970s, the western geopolitical argument about the advances of communism, the Domino Theory, was still cited, but it was gradually weakened by lack of popular support for the war and the loss of American blood and treasure invested in keeping communist influences out of southeast Asia. By the mid 1970’s, the US had pulled out of Viet Nam leaving behind millions of casualties and little to show for the effort. Added to Southeast Asia was the self-destructive meddling with the Cuban communist state. Castro died of old age in his communist bed.
There is an old saying that went “He’d complain if they hung him with a brand-new rope”. The suggestion was that some folks would complain about simply anything. Beyond the geopolitical and apparent military threat of the USSR beating the US into space were the much-ballyhooed technological benefits of the program. One of the oft-cited spin-offs was a Teflon coating for frying pans. It was an example that most citizens would understand and appreciate. Many incorrectly believed that NASA invented Teflon. Actually, Teflon was discovered unexpectedly in 1938 by the DuPont chemist Roy Plunkett.
Libertarians have argued (to my face) that if we wanted non-stick Teflon frying pans, why not just invent it? The introduction of any new and revolutionary product by a corporation carries financial risk and possible loss of reputation. The vaunted contribution of NASA to perfluorocarbon (PTFE) polymers was to push forward projects that required PTFE specifications. Industry did the rest by finely developing technology for PTFE production to meet the demand. Once there was an inkling of demand, industry kicked into gear. It was market-pull by that time.
NASA is very much in the technology-push world whereas many businesses are more safely oriented to market-pull. Technology-push is about invention of leading-edge vehicles, equipment, substances, instrumentation or services. Technology-push requires early adopters willing to wager that the new tech will give them a competitive edge. Government provides a ready-made early adopter.
Market-pull is where a manufacturer produces known or existing products and services. They compete by offering better availability, price and quality than their competitors.
Technology-push is the world of the tech startup. A start-up founder has a product or service that is sure to be a hit if only their products could get manufactured and pushed into the market. Tech investors will examine the startup’s business and financial plans and take a closer look at the technology or service to be offered. Is there a prototype? Is valuable intellectual property protected under patent? How stable is the supply chain or is there one? Will the company be sustained on the tech product only or will consumables be produced as well.
Importantly, is the technology-push startup looking to produce just a single product or is the technology expandible across a spectrum of applications? What if the product performs below acceptable tolerances or simply fails in the field? A startup with everything invested in a single product model is a “One-Act Pony”. Wonderful though the One-Act Pony may be, it can get sick and die in the marketplace. It can grow old and obsolete, giving way to falling sales and the mad scramble to develop a replacement product. I’ve been a part of 2 startups hoping to produce one-act ponies. The ponies died and we hit the streets.
Investors can analyze market-pull business plans by looking at the economics of demand as well as distribution of existing or similar products. Annual sales can be estimated, EBITDAs calculated, and profit margins uncovered. If the profit picture fits the general business model and timeline of the investors, they can release funding rounds to the startup with benchmarks to be met.
Necessity as the mother of invention?
In normal circumstances, industry operated by ambitious people may be motivated to advance their technology skillset to realize entry into new and promising markets. However, that said, an industry that only acts to match technological advances set by competitors is not showing the mettle required to launch a new paradigm in the technology-push manifold. Merely matching the competition does not quite describe a technology-pusher.
A technology-pusher is likely to find that they must walk the manufacturing highwire without a net and perhaps for a long time. Unless you are quite wealthy, launching a startup will likely hold personal financial risk. Commonly, external funding means that some percentage of ownership or shares will be given to the investors. By the time the product or service hits the market, the founders may find themselves as minority stockholders. Their dreams of grand wealth and influence is tempered by reality.
A naïve book-end view of technology pushers. Scientists are by nature more interested in phenomenology and naturally may see a two-dimensional universe of space and time. Scientists may gravitate to precision and accuracy while the engineer is also interested in not just precision and accuracy but also costs. When developing an engineering design, the engineers will constantly consider costs within the boundaries of space and time. Graphics by Arnold Ziffel.
A technology-push company is often started by engineers or scientists with experience in a particular subfield. Scientists commonly receive little or no business education as a degree requirement. Their role is the science guru. Engineers, on the other hand, fully understand the cost imperatives of a project and are able to design to remain within tight cost constraints.
In science, scientists are the main honchos. In business, engineers are the princes of the kingdom. They design projects, lead them, and come in on budget on time. A CEO with an engineering background is not at all unusual. They understand money part.




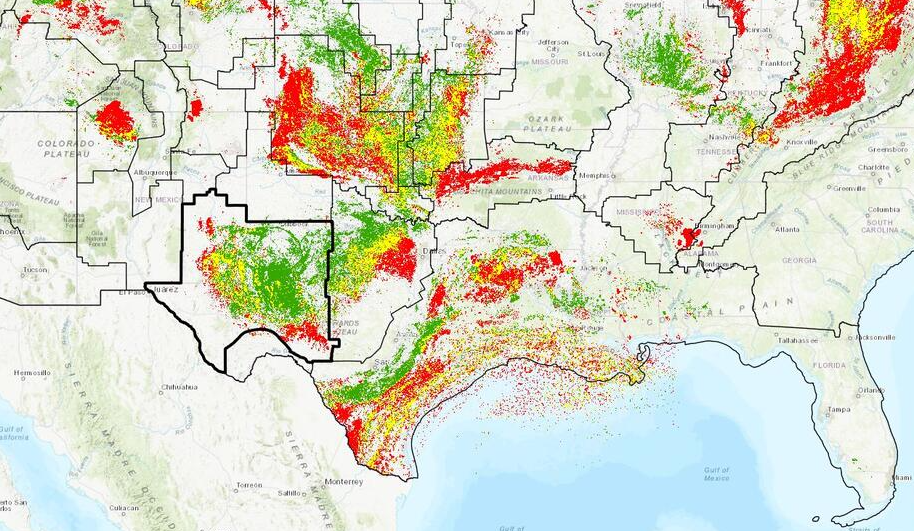
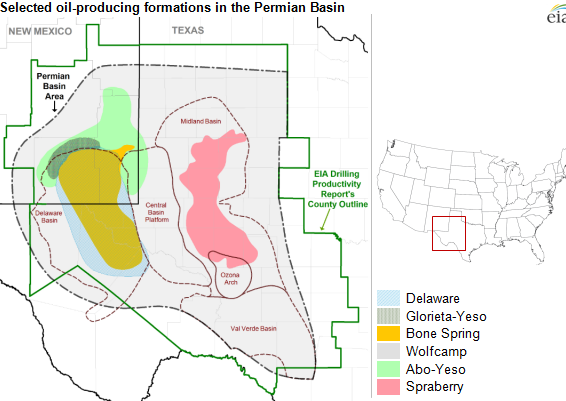

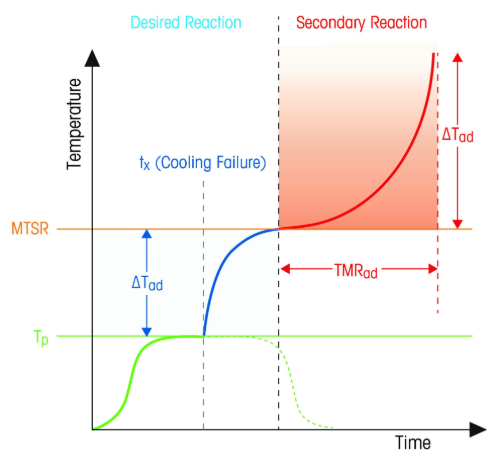

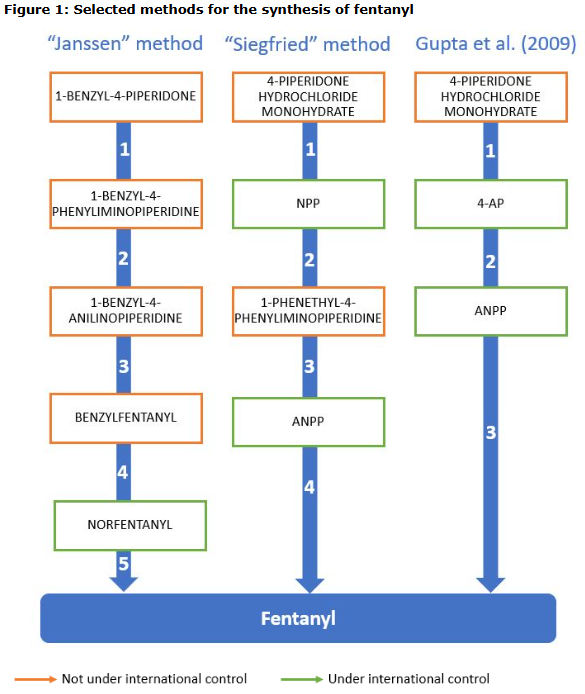

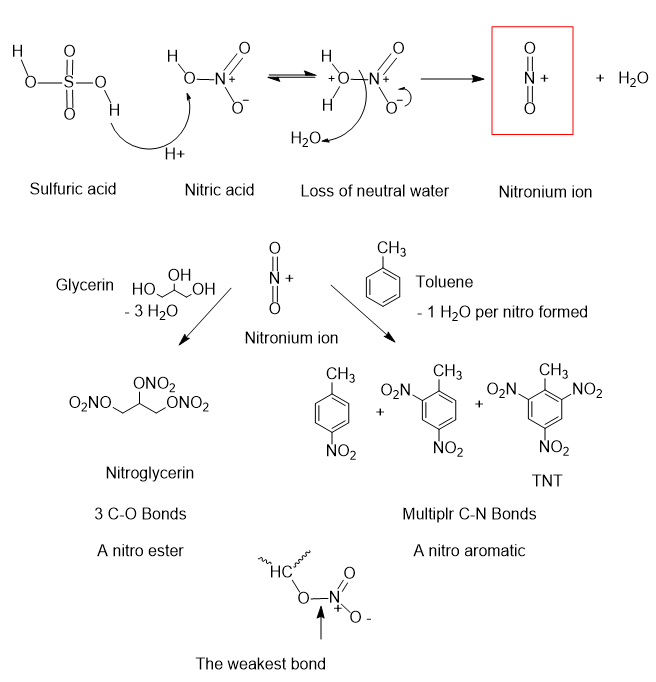
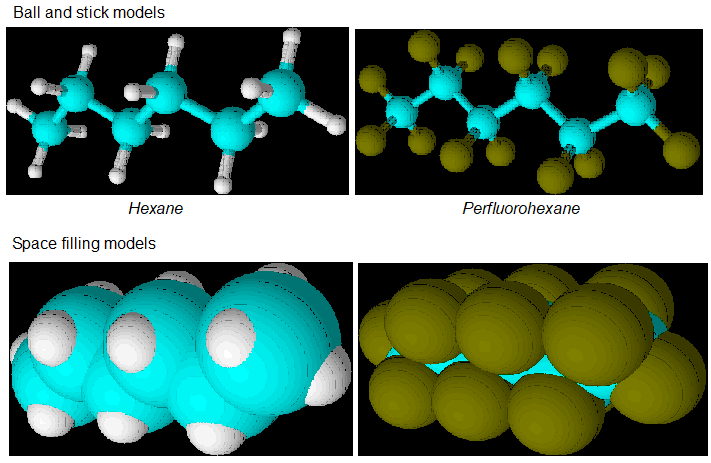
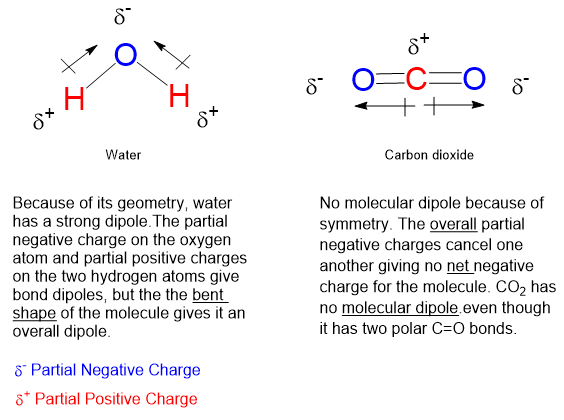


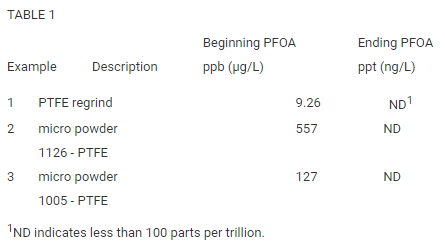
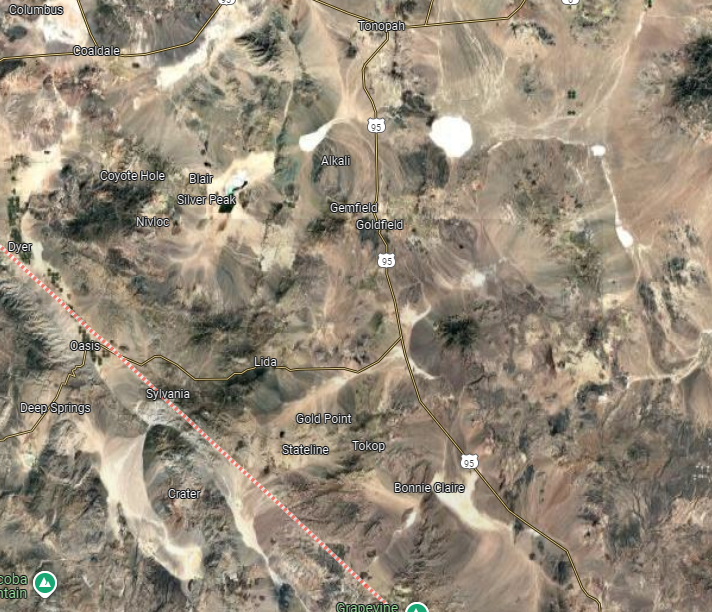
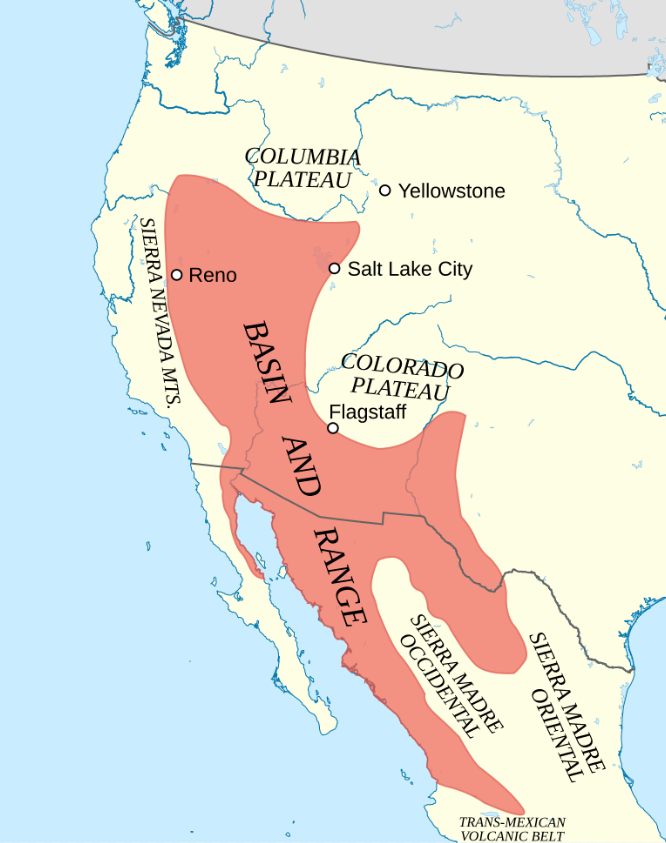

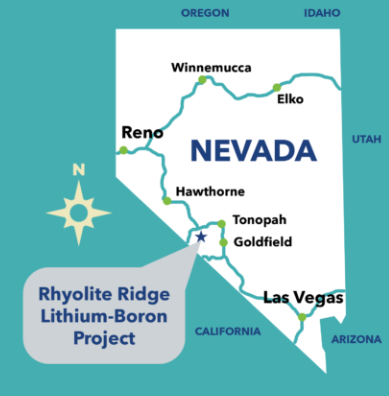

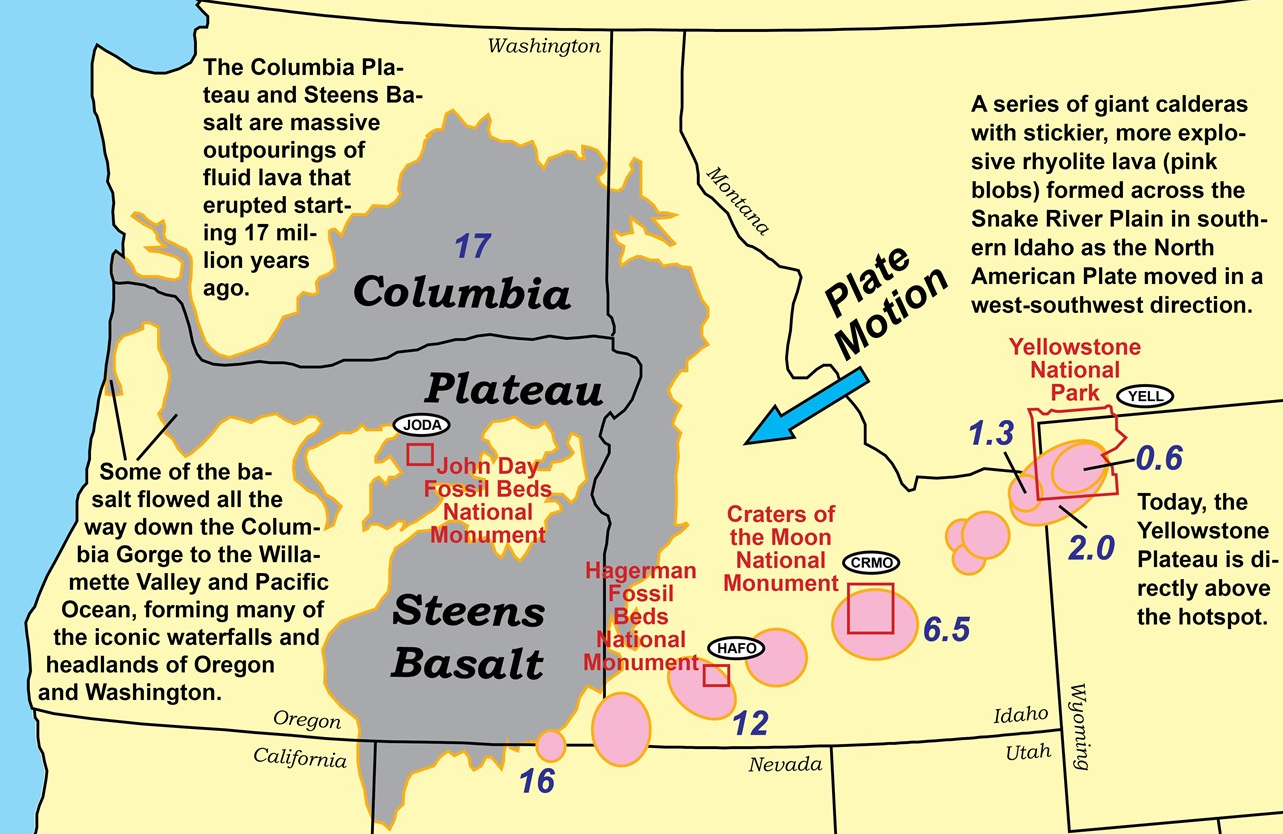

{kind=link}