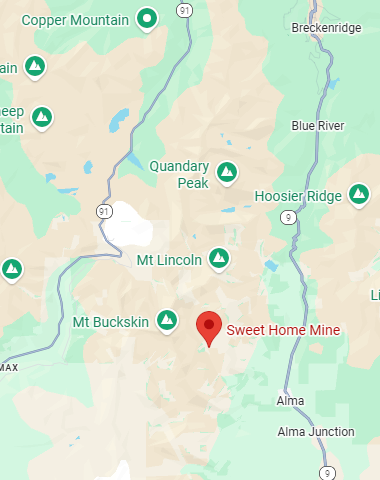
Not a single reader has asked about the photograph in the header of this blog, so I’ll save the many peoples of the world from having to ask. Mineral collecting has been a lifelong weakness of mine so there was no surprise when I bought the pink mineral in a rock shop in Leadville, Colorado. The pinkish mineral in the sample is rhodochrosite, the state mineral of Colorado. Like most samples, it comes from the now-closed Sweet Home Mine, a failed silver mine in Buckskin Gulch outside of Alma, CO, between Breckenridge and Fairplay. If you are ever in Denver with spare time on your hands, the mineral collection at the Denver Museum of Nature & Science has a stunning collection on display of rhodochrosite from the Sweet Home Mine.
Source: Google Maps. Location of Sweet Home Mine outside of Alma, Colorado.
To get to the site take gravel road 8 from Alma up Buckskin Gulch which eventually terminates at a trailhead near the base of several fourteeners in the Mosquito Range. We once tried to find the mine by driving up the gulch above Alma, but there were no signs identifying the mine.
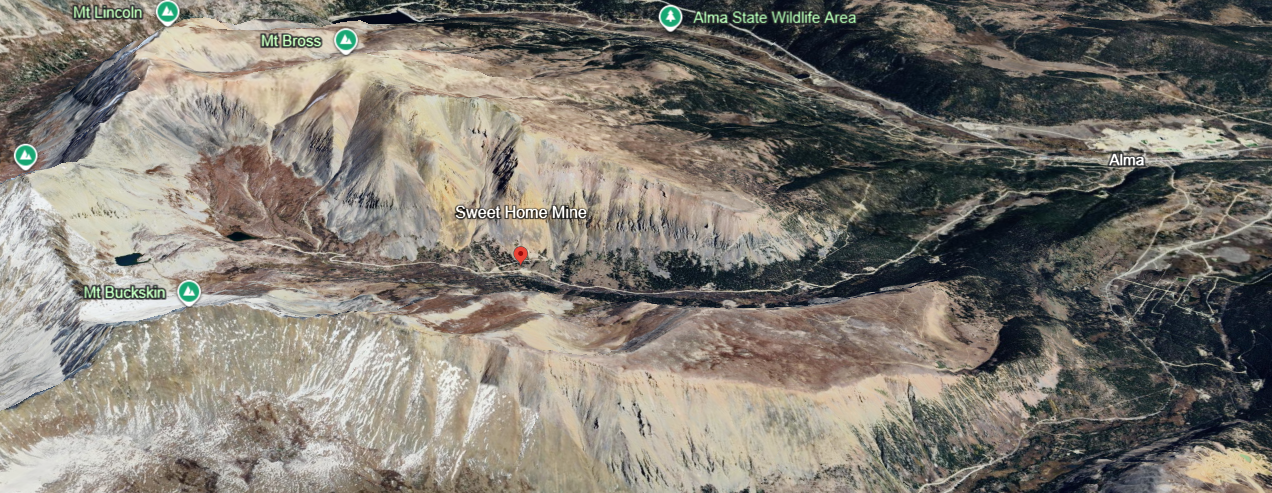
Source: Google earth. Location of Sweet Home Mine in Buckhorn Canyon.
While we did not positively identify the mine on our trip, a photograph (below) was found later of a building associated with the mine. We did see it but sailed right on by. The mine is located on private property so wandering around the site is not permitted.
Source: Facebook. The famous Alma King rhodochrosite specimen with museum dudes for scale.
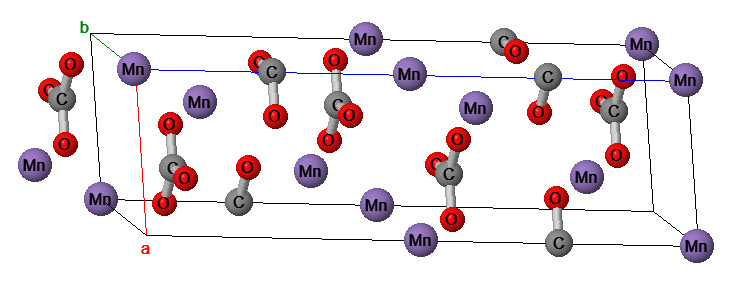
Source: personal specimen purchased at a rock shop in Leadville, CO. The rhodochrosite section is placed next to manganese on the periodic table just because it looked cool. The gold-colored bits on the specimen are likely chalcopyrite.
The mining district was discovered in the usual way- the search for placer metals like gold led miners up Buckskin Creek into the gulch looking for the source of the lode deposit. Originally a silver mining claim was made in 1873. The sporadic silver mining operation was abandoned in 1966. In 1991 the mine was bought out by Collector’s Edge Minerals, a consortium, and modernized. After a period of activity, the Sweet Home Mine was closed in 2004. However, another mine called the Detroit City Portal was begun by Collectors Edge on nearby Mt. Bross in 2016. This new operation, yielding many fine specimens was finally closed in September of 2024.
Source: Mindat.org. Looking north towards the Sweet Home Mine and what appears to be Mt Democrat on horizon.
“Mineralization is generally in base metal-silver-rhodochrosite-fluorite veins predominately hosted by meta-igneous and metamorphic rocks, with minor mineralization in porphyritic dikes and pegmatites. There are five main veins in descending order of production: the Main, Tetrahedrite, Watercourse, Blaine and Blue Mud veins. The Blue Mud Vein is a barren post-mineralization fault-vein, and production from the Blaine Vein was minor. Overall, the planned extent of the mine is small (1000 feet x 400 feet) with about 5,000 feet of workings, and the overall hydrothermal alteration zone small, despite evidence of on-strike continuation of the veins in the collapsed Tanner Boy workings directly across Buckskin Gulch. And even within a vein, rhodochrosite finds were limited.”
“Three conditions were responsible for the formation of vugs: (1) changes in strike and dip of veins, (2) vein intersections, and (3) openings formed by fault bends controlled by host rock foliation. In general, the 2nd condition was responsible for major pockets, and the 3rd for most smaller pockets. Exploration focused on fault/vein intersections. Fluid inclusion studies suggest that the hottest fluid flow produced the gemmiest ruby-red rhodochrosites.” Minedat.org
Deposits found in the mine result from mineral-saturated hydrothermal fluids moving from the mineral source-rock into faults and fractures in the formation that were cooler, leading to precipitation of the minerals. The large size of the rhodochrosite crystals in the museum collection suggests that the precipitation was gradual.
According to Minedat.org, after the buyout of the Sweet Home Mine by Collector’s Edge Minerals and subsequent modernization, ground penetrating radar was used to survey for vugs. According to the AI overview by Google in a search for “vugs”-
Vugs are- “small to medium-sized hollow spaces or cavities within rocks, often lined with beautiful, well-formed crystals like quartz or calcite, formed by mineral-rich fluids filling natural voids left by dissolution, tectonic shifts, or gas bubbles in volcanic rocks, prized by collectors for their exposed crystal formations.”
Only makes sense, right? Liquids within voids in the rock have the opportunity for crystals to grow into. Vugs are associated with faults and fractures which can be filled with hydrothermal fluids within a formation. Lode gold, silver, lead, etc., as well as quartz may line or even fill the vug. This is why some of the best mineral crystals are only found in mines and this certainly applies to rhodochrosite. Rhodochrosite contains manganese (II) which is oxidizable to a higher, more positive oxidation state, so protection from atmospheric oxygen deep within a rock formation prevents decomposition of the mineral.
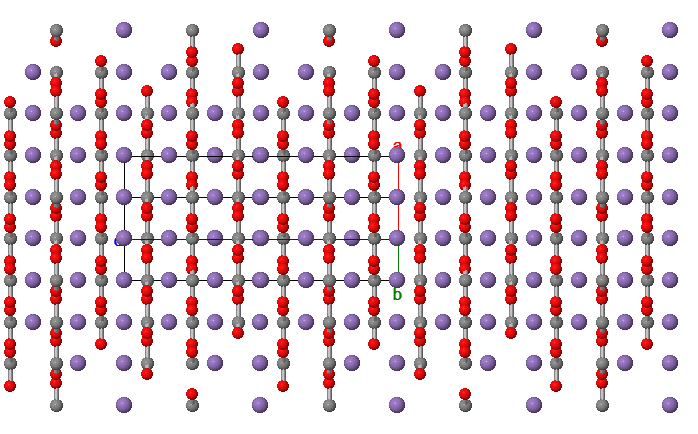
Crystallographic structures of rhodochrosite are shown below-
Source: Mindat.org. A view of the crystal structure rotated to see the planar arrangement of Manganese (2+) in purple and carbonate anions (2-) in grey and red.
Source: Minedat.org. In this view the alternating layers of carbonate anions (CO3 2-) delineating the carbon and oxygen atoms. The trigonal shape of carbonate can be seen.
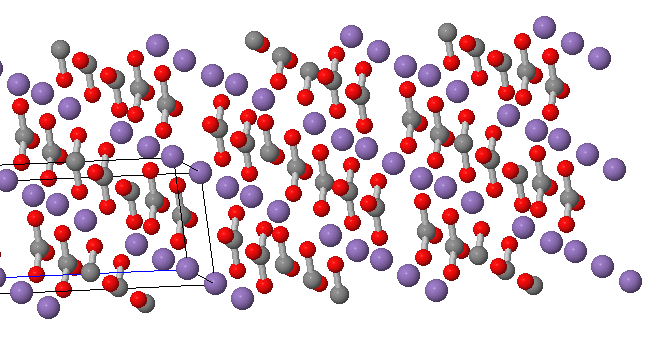
Below is a representation of the unit cell with atom labels. Clear images are tricky with crystal structures. Overlapping features are hard to avoid.
Source: Minesdat.org. The labeled unit cell of rhodochrosite. Partial carbonate structures can be seen contributing to the unit cell.
Rhodochrosite is manganese (II) carbonate, MnCO3, and is insoluble in water but as a metal carbonate it is acid sensitive and therefore subject hydrolysis or chemical or microbial oxidation to Mn(III) or Mn(IV). Like a great many common ionic substances, it is not regarded as suitable for jewelry applications because it is not comprised of silicate or aluminum silicate subunits common in semiprecious and more robust minerals like sapphire, beryl or garnet. The structure is composed of MnO6 octahedra connected by trigonal carbonate units. The large buff-colored balls are manganese atoms and the smaller, bluish-colored balls connected directly to the manganese atoms are oxygen atoms. The middle-sized darker balls not connected directly to the manganese atoms are the carbon atoms of carbonate.
Manganese is not uncommon in the Colorado Rockies. A mining geologist once complained to me that there was so much manganese in their gold mine tailings that it was a regulatory problem for them. For a time pyrolusite, or manganese dioxide (MnO2), was mined in Colorado, near Salida. Never a large operation, pyrolusite could be used in the extraction of gold from its ore.
Crushed pyrolusite was placed below a wooden container along with sodium chloride. To this mixture was added concentrated sulfuric acid. This generated gaseous hydrochloric acid which was then oxidized by the manganese dioxide in the pyrolusite into chlorine gas which flowed up through the container of gold ore combined with the gold ore and generated gold chloride. The water-soluble gold chloride was removed with water, then isolated and into this pregnant solution was dumped scrap iron. The iron reduced the gold chloride and finely divided gold precipitated out. This was a pretty danged clever method for use in the field as it required only water, NaCl, H2SO4 and pyrolusite mineral which could have been mined in Colorado.
Oh, BTW. You might know that a way to generate a stream of fairly dry HCl gas (in a lab fume hood!!!) is to place granular NaCl into a vented flask and slowly drip conc H2SO4 from an addition funnel on it. A stream of nitrogen is used to force a flow of HCl out of the flask and through a sparge tube into your reaction flask.
And, speaking of metals ...
Nearby the Sweet Home Mine, a hop, skip and a jump across the ridge to the NW is the Climax Molybdenum Mine on Fremont Pass just west up the road from the Copper Mountain Ski Resort. This major mining operation is owned and operated by Climax Molybdenum Company, a subsidiary of Freeport-McMoRan. If you look at the image for a minute, perhaps you can see that most of Bartlett Mountain is gone. Just imagine laboring in a frigid mine above the 11,000 ft altitude. I’d be dead by noon the first day …
Source: Google Earth. Just a few miles NNW of the Sweet Home Mine is the Climax Molybdenum Mine on Freemont pass.
The mineral of interest at the Climax is molybdenite, or molybdenum sulfide, MoS2. The deposit was discovered in 1879 by prospector Charles Senter who was actually prospecting for gold or silver. By 1895 Senter found a chemist who determined that the mineral contained molybdenum. At that time, however, there was no market for the moly. In a few years steelmakers discovered that molybdenum had application in steel making and, with the onset of WWI. the mine went into full production after it was discovered that the Germans were using it to strengthen steel in their tanks and weapons.
The National Mining Museum and Hall of Fame down the road in Leadville has a large collection of interesting artifacts from early mining efforts at Climax. If you have been in many mines, you’ll know that they are mostly hallways that have been blasted out of solid rock. When mining activity stops, they are eerily quiet.
Image source: National Mining Museum in Leadville, CO. Colorized photo of lunch time in the mine.
Molybdenum sulfide is also valued as a dry lubricant for use in the temperature extremes and vacuum of space. Dry, low vapor pressure lubricants are used to prevent evaporation and contamination of optical surfaces on a satellite.
This is an encore release of a much earlier post. –gaussling
After an insane week in the lab a road trip to the cool meadows of the nearby mountain range was just what the doctor called for. It was the last weekend before the family- one teacher and one kid- return to school. Summer break 2009 is history.
We piled in the car and pointed it uphill towards Leadville, Colorado. The planetary atmosphere thinly blankets this insanely high mountain city. It was just what I needed to clear my scrambled mind. Nothing like blinding sunshine and mild oxygen starvation to reset a brain in chronic spasm from sensory overload.
Leadville sits at 10,152 feet above sea level. If you doubt the effect on your stamina, just take a short sprint in any direction. Or just plod up the stairs of your hotel. Lordy. All of those business dinners- all that lovely Cabernet and Crème Brûlée- and years of driving a desk have caught up with me.
Leadville is located in the Colorado mineral belt and began to populate with fortune seekers about the time of the Colorado gold rush in 1859. Some placer gold was found in the streams, particularly in what was then called California Gulch, but for the most part Leadville became a silver camp.
In 1874, two investors with metallurgical training, Alvinius B. Woods and William H. Stevens arrived in Leadville and analyzed the muds found in the local sluicing operations. According to A Companion to the American West, edited by William Francis Deverell, (2004, Blackwell Publishing, ISBN 0-631-21357-0, p. 319) Woods and Stevens found the heavy black mud so problematic for gold sluicing was in fact composed of lead carbonate with high levels of silver. Woods and Stevens invested $50,000, quietly buying as many claims as they could and began hydraulic mining operations immediately.
By 1890 there were nearly 90 mines in operation employing 6000 miners. At its peak there were 14 smelter operations supporting the mines. Leadville was a genuine boom town with the expected mix of characters.
A mine is a hole in the ground with a liar standing at the top.
All mining towns have characters who go on to dominate local legends and stories. Among the well-known-for-being-famous rags to riches to rags players in Leadville are Horace and Agusta Tabor, along with Horace’s mistress and 2nd wife, Elizabeth “Baby Doe”.
To make a long story short, Horace was a struggling shop keeper who invested in a mine east of Leadville. Though it was salted by the previous owner to entice buyers, Tabor dug 25 ft further down the shaft and struck a rich and extensive vein of silver ore. The operation was called the Matchless Mine, after Tabor’s favorite brand of chewing tobacco.
According to the tour operators, Tabor operated the Matchless Mine 24/7 for 13 years, pulling an average of $2000/day of silver out of it. At its peak, the mine is said to have employed 100 people. Miners were paid the common rate of $3.00 per day to climb 365 ft to the bottom of the shaft for 12 hour shifts.
The underground workings of the mine followed the vein structure and focused on sending concentrated ore to the surface. Buckets carrying approximately one ton of ore per load (my estimate) were tipped into ore carts and rolled into the ore house for hand sorting. The most highly concentrated and valuable ore was dumped down a chute for loading into a rail car and the gangue (or tailings) was dumped into the gulch.
An assay building (not shown) was on site to provide a continuous assay and accounting of silver sent to the smelter in Pueblo, Colorado. Unlike many other mine operators, Tabor owned a rail operation and had a spur at the mine for pickup and delivery of ore. Many mine operators had to employ mule-skinners to cart wagon loads of ore to a rail siding for transport to the nearest smelter.
In 1893 the repeal of the Sherman Silver Purchase Act and the collapse of the railroad industry bubble were part of a panic that lead to a crash in silver prices. Tabor lost everything and, as a respected public figure, was appointed postmaster of Denver for a short time. Eventually Tabor died at age 69 in 1899. Ex-wife Agusta had invested her divorce settlement wisely in Denver and lived comfortably. Widow Baby Doe Tabor was found frozen stiff in her shack at the Matchless Mine in 1935.
Matchless Mine Shack
All of the digging from the boom time of Leadville has left an enduring legacy for those who live in the watershed. Much of the mining activity occurred uphill, east of the city and as a result, that area is pock marked with many large colorful tailings heaps. While the colors are interesting to ponder and sample, the ground and surface waters are greatly affected by aqueous extraction of metals from these piles.
If you stand next to one of these heaps, you can’t help but notice the smell of sulfur. The ore and tailings are enriched in sulfides and once exposed to air and water, oxidation occurs to make corrosive runoff. This is a kind of heap leaching phenomenon that will eventually exhaust itself, but only at the cost of water quality.
Boomtown Legacy (Copyright 2009 All rights reserved)
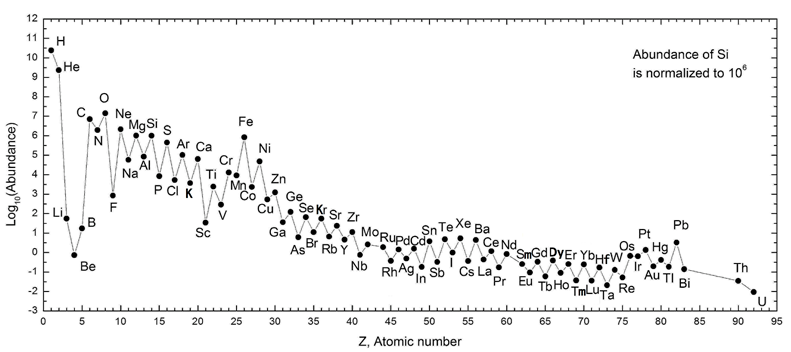
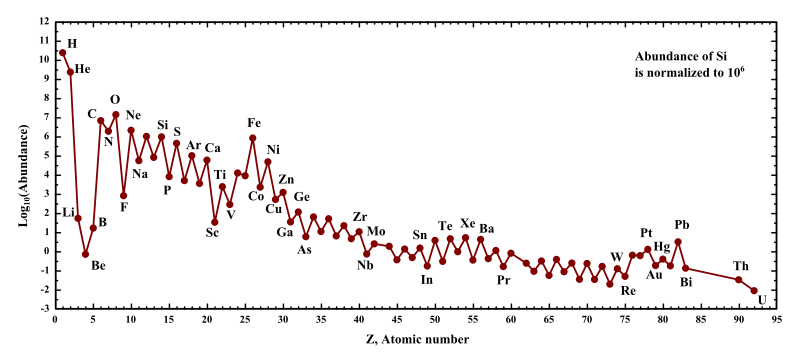
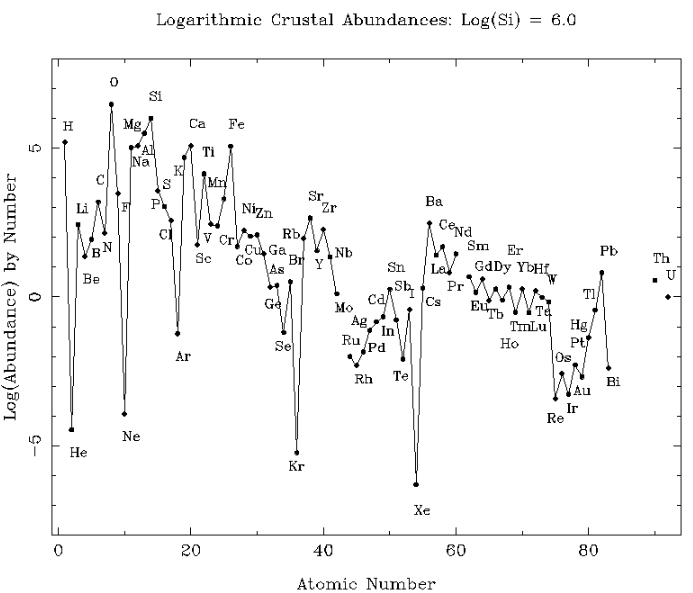
If you look the chart of elemental abundances in the cosmos below, you’ll notice that beyond hydrogen and helium atomic numbers, there is a steep log-scale drop in the abundances of lithium, beryllium and boron, or just ‘LiBeB’. This is followed immediately by a sharp exponential rise in the abundance of carbon, nitrogen and oxygen, etc. Beyond oxygen there is a general downward trend in elemental abundances. Well, except for iron, Fe. It is a special case related to the demise of stars. The zig-zag in the curve is explained by the Oddo-Harkins rule which postulates that even numbered atomic numbers are more abundant. I’ll leave this to the reader to explore.
Maybe 15 years back I decided to understand why each of the LiBeB elements are so scarce. And, if they are so bloody scarce, then how do they end up concentrated in ore bodies on earth? As a first-order approximation, you’d think that the explosion of stars and the resulting rapid dispersal of matter into the surrounding space would argue against the LiBeB concentrations on a planetary body like earth.
So, here is the question: Given that the cosmic abundances of lithium, beryllium and boron (LiBeB) are dramatically smaller than the succeeding light elements, how is it that “concentrated” ore bodies containing these elements exist on Earth?
Source: Wikipedia. “Oddo–Harkins rule.” Notice that the vertical axis is a log scale.
As I look at it now, the answer is bloody obvious. But when I asked the question 15 years ago, my understanding of hydrothermal activity and fluid movement through the earth’s crust was pretty slim nonexistent. But hey, my focus in college was not astronomy or geology.
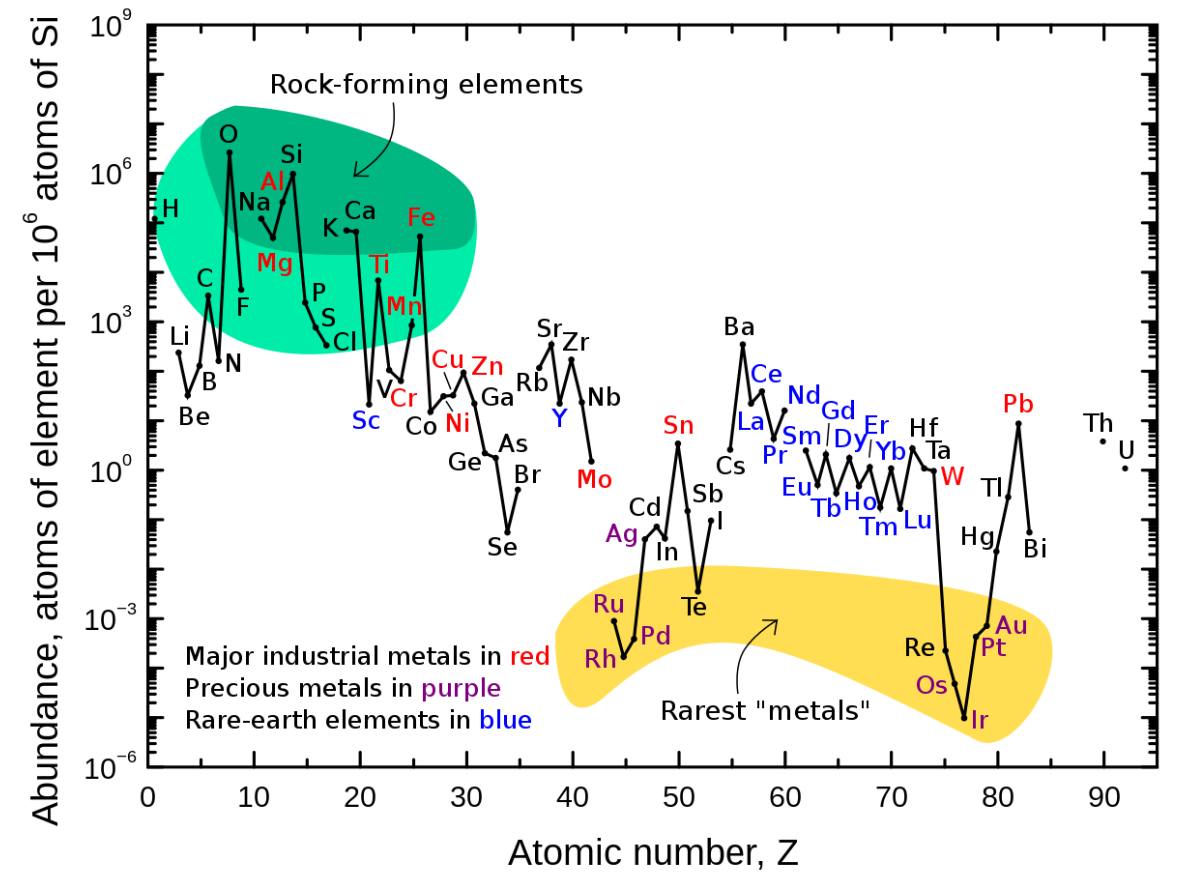
The graphic below shows how elements can be grouped according to a few particular categories. It is an absolute tragedy that the Platinum Group Metals (yellow shading) are in such the low abundance. These elements are uniquely valuable in chemical and industrial applications.
An even more fundamental question is, how is it that the cosmic abundances of lithium, beryllium, and boron are so low? Answer: they can’t survive the temperature and pressure conditions in the core of a star. They are fuels and therefore too delicate.
So LiBeB nuclei are produced during cataclysmic stellar explosions by some kind of spallation process, scattering fragments of larger nuclei into space. Another theory claims that cosmic radiation is responsible for the spallation of larger nuclei leading to LiBeB fragments. Subsequent generations of star formation resulting from an accumulation of hydrogen, helium, and heavier elements can lead to a star system with a protoplanetary disc where mass is brought into closer proximity. Mutual gravitational attraction between dust, chunks and aggregations of solid phase matter not already drawn into the star begin over time to aggregate and form planetary bodies. Some of the larger bodies are heated to liquid phase by collisions. Smaller mass objects may remain in the solid phase. Planet or moon sized bodies can collide to produce another planetary body or just debris. A fluid body of sufficient mass will spontaneously alter its shape in the direction of spherical such that all of the mass is as close to the center of gravity as possible. A spherical planet is one in which all of the mass is as close to the center of gravity with minimum potential energy as possible. A flat, disk-shaped body does not. It is hard to say just exactly how a flat planet-sized body would form with only gravity to drive it.
A cooling but still partially molten planetary body will begin to sort its large-scale composition by density and melting point. High melting point materials near a cooler surface will form solid crystalline bodies within the magma and then settle and possibly stratify according to density, then re-melt and disperse and convect to repeat the process. With cooling, the magma becomes increasingly viscous which would be expected to slow down mixing within the magma body. Over a long period, the planetary surface will continue to cool by loss of radiant energy and eventually the surface will crust over. Planetary bodies will accumulate mass by gravitational attraction as they sweep through space in their orbits, adding whatever chemical diversity that may be falling inwards to the surface. It seems reasonable to suppose that infalling dusts, rocks and asteroids are not uniform in their total compositions and may result in a non-uniform, spotty distribution of elements on the planet.
Solidification
Well before the surface cools to the point where a gas such as water can condense to form bodies of liquid water, water vapor in the atmosphere can convect to produce clouds at altitude, releasing latent heat and further adding to the heat transfer from the planet. Eventually, liquid water on the surface of a cooling planet can carry heat energy upward by conduction from the surface and atmospheric convection upwards, releasing the latent heat of condensation as clouds form in the upper atmosphere where it is colder. Heat transferred to the atmosphere will radiate infrared energy in all directions including space. The planet has developed weather.
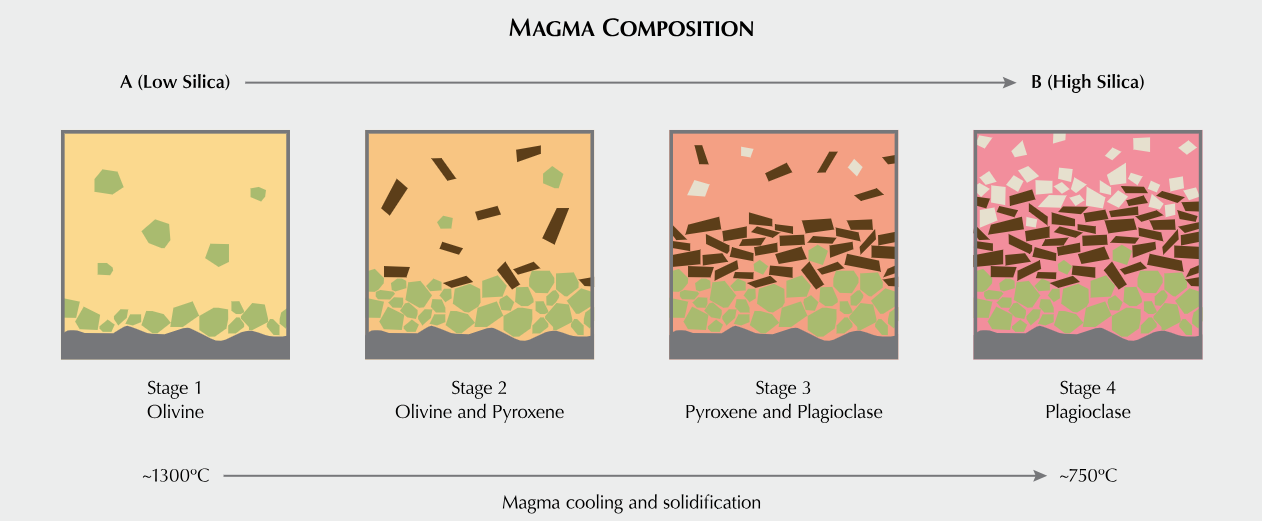
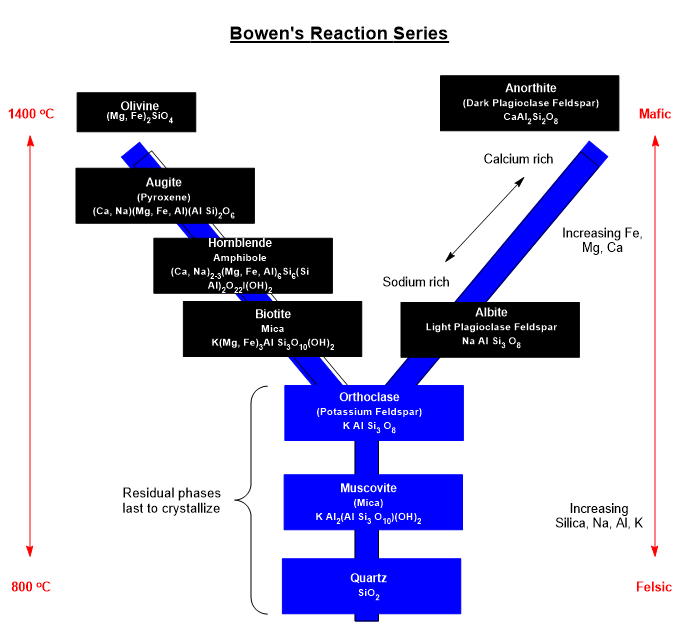
Source. GIA. Formation of minerals from magma. Minerals with the highest melting point begin to crystallize first, depleting the molten phase of those components. This is fractional crystallization described in Bowen’s Reaction Series.
Solidification processes in a magma begin to spontaneously partition or nucleate into particular combinations of elements. Some groups elements like silicates combine with metals into molecules (covalent bonds and ion pairs). For a period of time on any given crystalline surface, the temperature will support equilibration to and from the magma wherein ionic species will attach to the crystalline surface from the magma. With sufficient temperature, ions can detach from the surface and diffuse into the magma. This process will continue as the temperature drops freezing the magma and suppressing diffusion.
Bowen’s Reaction Series summarizes trends in the order of melting/freezing of various classes of minerals with temperature in magma. From location to location, the composition of magma is variable where some may be silicate rich and others silicate poor. That melting/freezing points of minerals vary with composition shouldn’t surprise my chemist friends.
Graphic: Shamelessly and laboriously redrawn by Sam Hill.
Reaction selectivity is driven by the sign and magnitude of Gibbs energy in a particular transformation and the ability of the combination process to be reversed at the local temperature. The formation of crystals or their dissolution in hydrothermal fluids or magma is taken to be a type of reaction. Many of the species present at magma temperatures will be ion pairs.
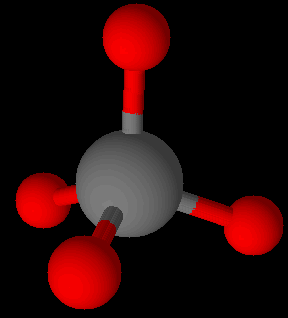
A few covalent species like the highly stable Si-O bonds in silicates are found in great abundance. The O of Si-O may or may not be connected to other silicon atoms such as … -O-Si-O-Si-O-Si-O-Si-O- … for examples. There is no upper limit to the number of O-Si groups possible in minerals, though their assembly can go many directions. Quartz is a 3-dimensional network homopolymer. All 4 of the oxygens on silicon are connected to adjacent silicon atoms via the oxygen linkages. Quartz is a mineral comprised of all covalent bonds. Quartz has a much higher softening temperature than manufactured glass.
Silicon readily forms tetrahedral connections with oxygen as in silicate (SiO4)4- units. Si-Si bonds are not found in nature.
In a hydrothermal fluid solution or in magma, ionic species randomly diffuse, collide or swap partners. Charged species like iron (2+) cations can collide but will bounce apart owing to their like positive charges. But at a sufficiently low temperature, a sulfide (2-) anion can collide with the oppositely charged iron (2+) cation and remain connected as iron sulfide. The two ions are now chemically bonded and in doing so, heat is released and dispersed into the surroundings leading to a localized increase in temperature. Heat spontaneously flows from high temperature material to lower temperature material. The localized heated surrounding crystallizing minerals can keep dispersing the energy outwards, but as it encounters more mass which also absorbs heat energy, the resulting temperature rise diminishes rapidly as the energy is diluted over more mass.
Chemical transformations have two broad drivers- kinetic and thermodynamic. Kinetically driven transformations favor the pathways that are the fastest Thermodynamic transformations are equilibrium-controlled meaning that transformations that are reversable will favor the end state having the lower Gibbs energy. The Gibbs energy (ΔG) combines the heat of reaction (enthalpy, H) minus the entropy (S) times the absolute temperature (T). The entropy accounts for whatever heat energy may be gained/lost from/to the environment. When a bond forming reaction occurs, heat is released and moves into the environment.
Mineral nucleation starts with a microscopic “seed crystal or compatible surface” that will provide a template to further crystal growth by like molecules. Some combinations of elements will begin to polymerize forming high melting point, low solubility molecules comprising silicates and aluminates.
The concentration of the Li or Be or B into what would later become economic ore bodies is driven by the flow of ground water. Subsurface hot water flow in the crust is referred to as hydrothermal flow and from it is where concentration of the LiBeB begins. An ore body is a mass or formation of rock that is enriched in some particular element or group of elements. Usually, these elements are part of chemical compounds rather than pure elements. Gold would be an exception.
Definition- Minerals and Rocks
Mineral– A solid substance with a well-defined chemical composition and characteristic crystal structure.
Water, and especially hot water under pressure, will chemically alter the rock it is in contact with. While it is in contact, some fraction of the altered rock will dissolve and some components may be entrained as suspended solids in the fluid. This will extract and partition part of the altered rock into a solution with a range of possible compositions depending on solubility, pH, temperature and pressure. In this way, elements get partitioned into solution phase and away from the solid phase.
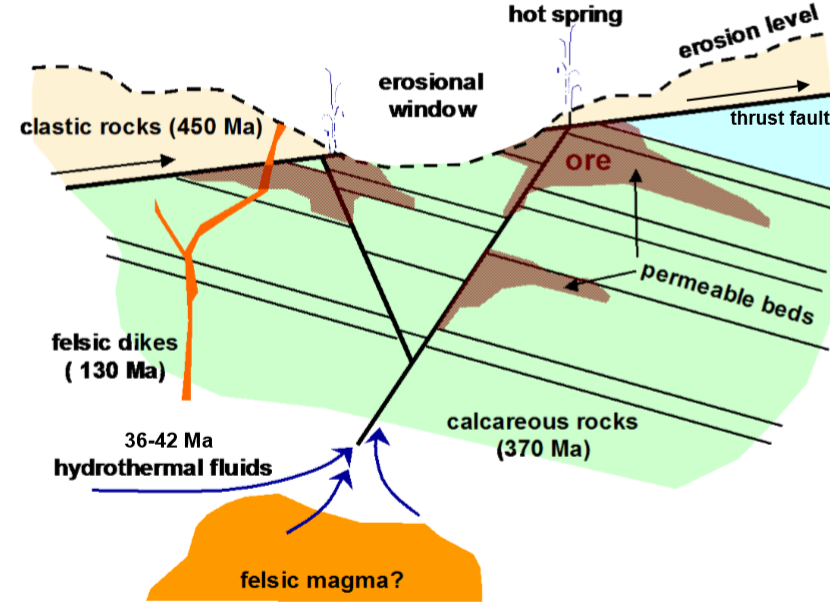
Source: Wikipedia. This diagram shows felsic magma as the heat and mineral source. Felsic magma is enriched in the lighter elements like silicon, oxygen, aluminum, sodium and potassium. Because of the enhanced presence of silicates and aluminates, the magma or lava tends to be more viscous.
Hydrothermal solutions can sit in place for a very long time, eventually saturating with mineral components. If there is cooling in place, the saturated solution can precipitate to form a solid body of mineral filling the local spaces. The deeper the fluid, the warmer it will be and likely the slower it will cool. However, if the surrounding rock is porous or develops fractures or faults, the hydrothermal fluid will flow to the zone of lower pressure. If the flow is into a cooler zone, there may be precipitation of the least soluble components leading to solid mineral formation. Precipitation can also occur from a drop in pressure.
Solubility vs Temperature vs Pressure Example with Quartz
The effect of temperature and pressure on precipitation of silicate can be seen with the beautiful pressure/temperature/solubility relationship shown below. For instance, we can see that for temperatures at or below 150 oC, there is very little change in solubility of silicate with increasing pressure. On the higher side of the temperature scale at 350 oC, we see that there is a considerable solubility change with increasing pressure. At high pressure, say 700 MPa, the solubility of silicate is most sensitive to changing temperature.
Source: For educational purposes only. Williams, Randolph; Fagereng, Åke Fagereng, The Role of Quartz Cementation in the Seismic Cycle: A Critical Review, March 2022 Reviews of Geophysics 60(1), DOI:10.1029/2021RG000768
The research article above cites the solubility of SiO2 in mg/kg water, or ppm. I am using the term silicate indicating hydrated and charged SiO2, the SiO4 tetrahedral form, which is known to dissolve in water. It is common in some of the geological literature to refer to the neutral oxide form of a mineral or metal.
SiO2 is often observed in its amorphous or crystalline topological polymer (or network polymer) form as a quartzvein, pegmatite or crystalline mineral component of a felsic igneous rock. Quartz found in a vein is there because a fault or fracture was available for filling with a hydrothermal fluid. As the curve above infers, saturation and precipitation can be abrupt with a sudden drop in pressure during a fault movement or sudden opening of a fracture network. Quartz veins are often accompanied by metallic gold in the upper oxidized zones (gossan) of the formation. In the context of mining, these veins are sometimes called quartz reefs.
Back to LiBeB
Although LiBeB elements are scarce, there are hydrothermal processes in the crust that can concentrate them in economic quantities.
An excursion into ore geology shows that selective hydrothermal extraction and transport are critical to the formation of a great many types of ore bodies. In fact, all three elements of LiBeB are moved by hydrothermal fluid transport on Earth at some point.
Lithium
Lithium metal is quite reactive and not found on Earth in the neutral metallic state. It is a single-electron donating Group 1 element and the lightest atomic weight metal of all the elements. Lithium has a large standard reduction potential of -3.05 Volts and is an excellent donor of electrons and a poor acceptor of electrons.
Lithium metal reacts with three major components of air: water, to form LiOH + H2; Oxygen to form Li2O + Li2O2 + H2; and nitrogen gas to form NLi3, lithium nitride. While other air-reactive metals can be stored under a hydrocarbon like kerosene for protection, lithium will float in these liquids and may be mixed with a heavy hydrocarbon or grease to keep it covered.
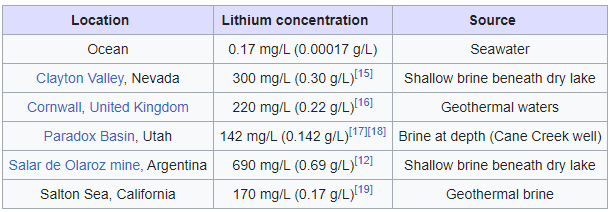
Lithium deposits can be split into two general domains- brines/muds/leachates, and hard rock deposits. Spodumene is the common hard rock source of lithium found in a few places around the world. Spodumene is lithium aluminum inosilicate and has a few variants such as Hiddenite, Kunzite, and Triphane coming from trace elements present in the surrounding rock.
The Silicate Zoo section is a bonus feature and may be skipped.
Bonus: A Step into the Zoo of Silicates
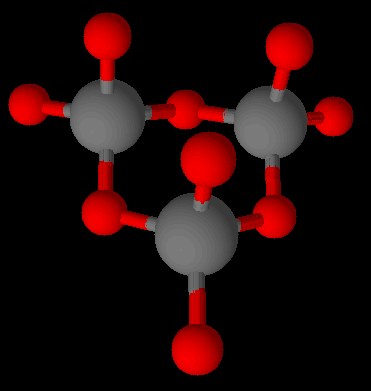
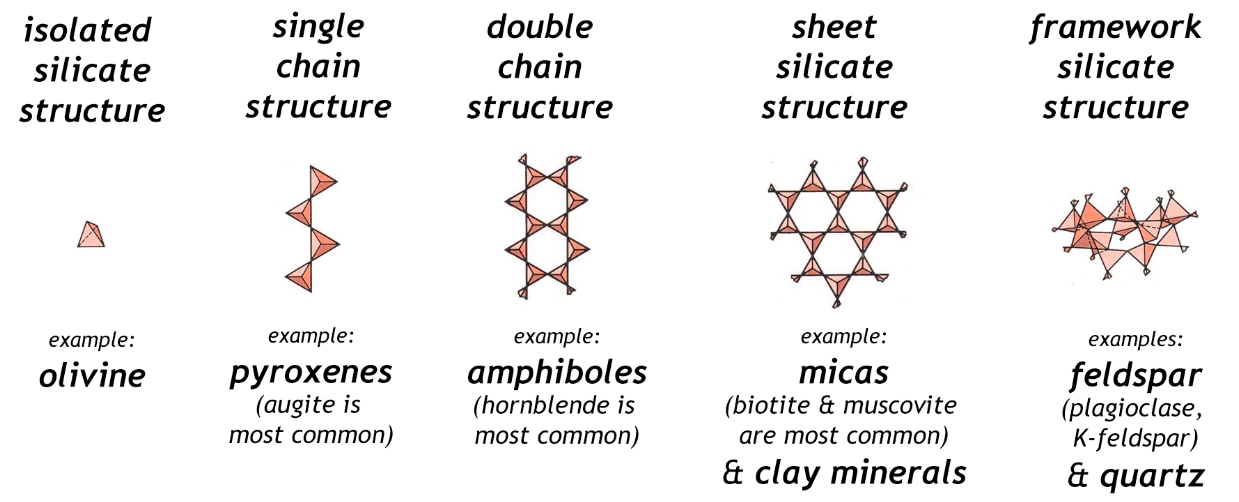
Spodumene is an inosilicate. Silicates as a group make up 90 % of the Earth’s crust. The basic unit is the tetrahedral [SiO4]4- silicate with 4 oxygen corners of the tetrahedron. These are known as ortho- or nesosilicates. The 4 minus charges are balanced by 4 plus charges provided by one or several metal cations at a time. Orthosilicates are the simplest variants of silicates.
A basic silicate ion, (SiO4)-4 with cations omitted. Graphics by Arnold Ziffel.
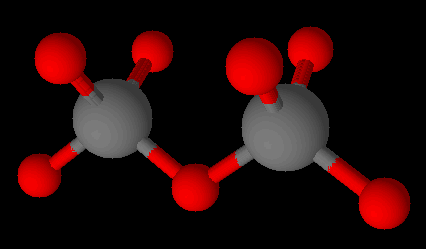
The corner oxygen ions of silicate can be attached to the silicon of another silicate. These are called the sorosilicates and have the formula [Si2O7]6−.
A sorosilicate ion, (Si2O7)-6 with cations omitted. Two silicates sharing an oxygen. Graphics by Arnold Ziffel.
Silicate units can also form rings called cyclosilicates. The rings consist of silicate tetrahedrons with alternating linkages of silicon-oxygen atoms. The general formula is [SinO3n]2n−.
A cyclosilicate, (Si3O9)-6 with cations omitted. Commonly called ‘D3’ when pendant oxygen atoms are replaced by CH3. Graphics by Arnold Ziffel.
There are several varieties of inosilicates. As a group they link silicate units into interlocking chains. There can be a single chain (pyroxenes such as spodumene) or two (amphiboles such as asbestos). The single chain inosilicates have the formula [SinO3n]2n−. The double chain inosilicates have the formula [Si4nO11n]6n−.
Monomeric silicates can polymerize along 1, 2 or 3 dimensions and by 1 or 2 connections to other tetrahedral silicates through the sharing of oxygen atoms. Graphic: Libretexts.
Phyllosilicates are sheet structures of silicate and have the general formula [Si2nO5n]2n−. This group includes micas and clays.
The tectosilicates are 3-dimensional frameworks and have the general formula [AlxSiyO(2x+2y)]x−. Note that aluminum may be present. This tectosilicate group includes quartz, feldspars and zeolites.
If you look at all of the general formulas, note that the negative charges must be balanced with positive ions (cations) of some kind. They are typically metal cations that vary in positive charge and ionic radius, although they can be capped off with hydrogen. A metal cation with its positive charge and ionic radius can be most easily replaced by a different metal with the same charge and similar ionic radius. Different charges and ionic radii are possible replacements but cause distortions in the lattice structure.
Minerals can form such that a silicate [SiO4]4- unit can be replaced by one or more aluminate (AlO4)-4 units and the metal counterions can be substituted by other metals of the same charge and similar ionic radius.
Back to Lithium
A growing fraction of lithium exploration and production involves lithium brines. Subsurface brines enriched in lithium (up to 0.14 %) can be pumped to the surface and exposed to sunlight in large evaporation ponds. Brines are saline solutions comprised of numerous soluble ionic substances. The lithium component must be isolated to a specified level of purity prior to sale. The most common product leaving a lithium mining operation is the insoluble lithium carbonate.
The process of concentrating a mineral from the ore is called beneficiation. An ore is comprised of the target mineral and gangue or mine waste from previous activity. The idea is to use economical physical and chemical methods to remove the gangue from the target mineral.
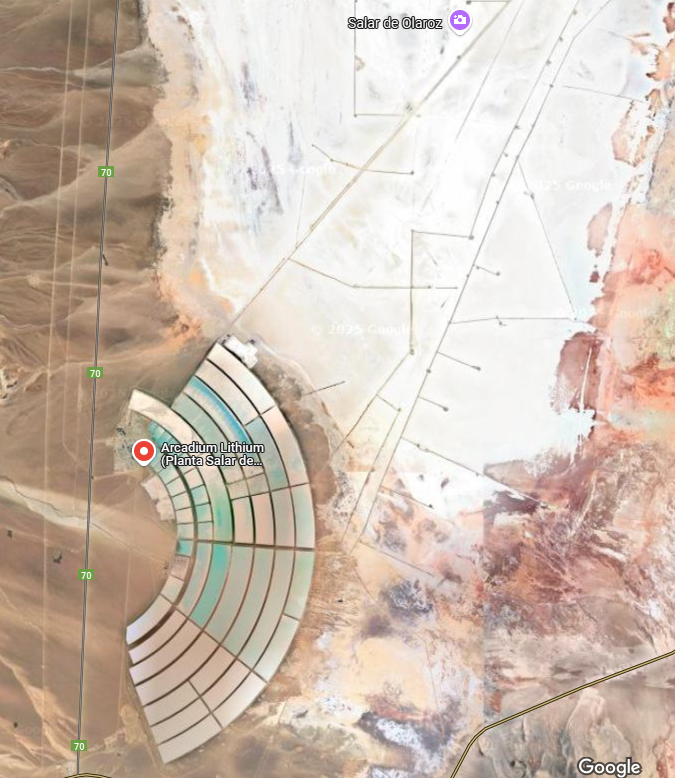
The largest collection of lithium brine reservoirs, known as salars, is in the Lithium Triangle in Latin America. The countries in the Triangle are Chili, Bolivia and Argentina. This area holds more than 75 % of the world’s lithium resources under their salt flats.
The salar brine is pumped into an evaporation pond and allowed to concentrate in the sunlight for a year +/-, then it is filtered and sent to another evaporation pond for further concentration. What happens next depends on which of the several possible processes is being used. The type of processing that is used depends on few things: The composition of the ore and interfering substances present; the company may have in-house technology they can adapt; there may be patent constraints in force; the process economics will apply considerable effects on equipment size and required annual throughput.
Source: Google maps. The Yanacocha gold mine owned by Newmont. The curved features are evaporation ponds taking advantage of evaporation and subsequent concentration of the brine by solar energy. This process is fractional crystallization, precipitating the least soluble ion-pairs in the presence of more soluble ion-pairs. An ion pair is defined as XmYn, where X is a cation of variable charge and Y is an anion where m and n are numbers such that m times the cationic charge plus n times the anionic charge add to zero net charge.
Economic ore is that resource which can be beneficiated and further refined and sold at a profit. So, the amount of ore that constitutes an economic deposit can shrink or grow with the market price of the product.
A recent development in lithium resources is the Rhyolite Ridge Lithium-Boron Mining Project near the ghost town of Rhyolite, Nevada, and southwest of Tonopah. The mine will be a large scale open pit operation operated by the Australian mining company ioneer. The mining will be carried out in the conventional drill-and-blast, and load-and-haul method. In the beginning the mining fleet will use automated haul trucks.
Even though nature has provided a concentrated lithium ore body, people still have to go to considerable lengths to produce economical lithium product, all the while gambling on the market price in the future.
Beryllium
Beryllium, Be, atomic number 4, has some very useful properties that make it a valuable metal. It is also quite scarce. The world’s richest beryllium deposit is located near Spor Mountain in Utah. The mine is operated by Materion Brush Inc., formerly known as Brush Wellman. The mine is operated by Materion Inc., formerly known as Brush Wellman. The beryllium rich tuff is located by trenching and drilling. Tuff is compacted volcanic ash that has been cemented into porous rock by contact with water flows. The area also contains fluorspar and uranium, separately, in combination with beryllium. Bertrandite (Be4Si2O7(OH)2) and beryl (Be3Al2Si6O18) are the chief beryllium-bearing minerals and fluorspar is a common accessory mineral.
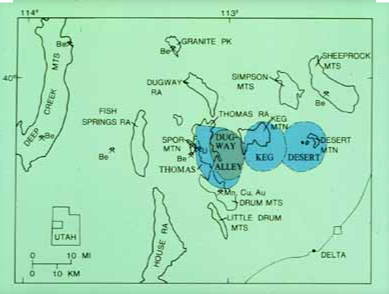
Spor Mountain is located in South Western Utah in basin & range country that extends across Nevada to California.
The above map is from the US Geological Survey and shows 3 extinct calderas in the immediate Spor Mountain vicinity represented in blue. It is interesting that tuff, made from pyroclastic flows and ash where particulates are later cemented together, is the rock in which beryllium is found in the area AND there are 3- count ’em 3 -calderas right nearby. Coincidence?
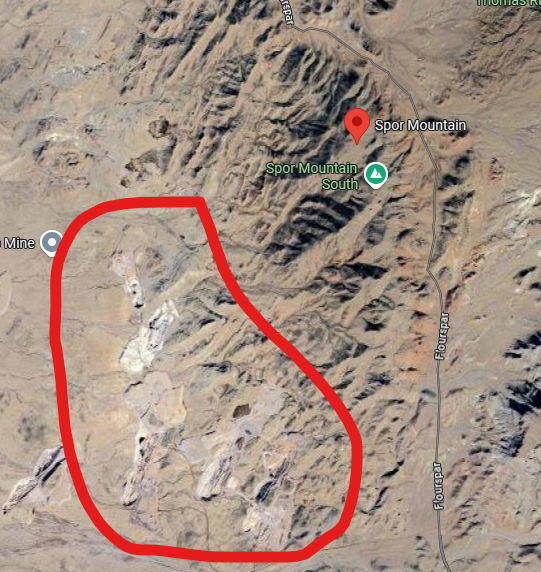
Source: Google Maps. The red circle shows the location of Spor Mountain beryllium mines in Utah. Closer examination of the mines shows that the mines and roads are bare of vehicles. Nearby, a parking lot with large haul trucks can be seen, but the overall location leaves the impression that excavation of the ore has paused.
Beryllium is a rare element on earth showing up in veins, pegmatites and tuff. In the cosmos its existence is thought to be due to cosmic ray spallation.
“The beryllium tuff is a favorable host for beryllium ore because 1) it is adjacent to faults and rhyolite vents where mineralizing fluids could enter the tuff, 2) it is a porous, reactive conduit for mineralizing fluids, including both hydrothermal and ground waters, and 3) it contains carbonate clasts, which reacted with fluorine-rich fluids to precipitate fluorite and beryllium.” Source: USGS, 1998
Source: USGS, 1998. A rare showing of Be ore at the surface. The white band runn9ng diagonally across the image is the ore body. Most Be mining at Spor Mountain is open pit mining.
Boron
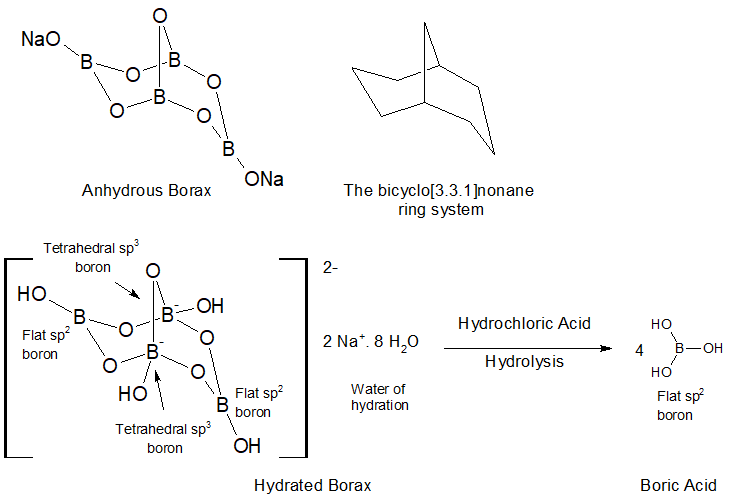
We’ll defer to the Wikipedia to provide background information on the element boron. The main source of economic boron is borax. Borax is a type of evaporite originating from hydrothermal fluids that have flowed to the surface where subsequently the water carrier evaporated to afford dissolved and suspended solid borax, a hydrated borate. Boron the pure element is not found, it only appears as set of various borates. There are monoborates, nesoborates, inoborates, phylloborates and tektoborates. The prefixes below are applicable to silicates and other oxides as well. The older Nickel-Strunz categorization system has been used to describe the classification of complex borate anions found in minerals.
Hydrolysis of Borax to boric acid. Graphics by Arnold Ziffel.
The structures of borax and hydrated borax above have been reduced to their minimum hydration. In reality these are idealized structures not reflecting the agglomeration into larger hydrogen bonded structures. Neutral sp2 boron atoms have an empty, low-lying p-orbital which can bond to the O of O-H groups whether from water or other borates.
The actual composition of any oxy-borate including borax will depend on its thermal history and its chemical environment. Attempting to dehydrate a borate may indeed remove water, but in doing so may open up a p-orbital on boron that would allow an exchange of oxygen species, whether another borate, boric acid or just another water molecule. This is not worrisome when producing boric acid, but when one of the OH groups of boric acid is replaced with a carbon atom, then dimerization and trimerization may occur even at low temperature under vacuum to form boronic anhydrides.
When ordering an organoboronic acid, look at the specs very carefully. Sometimes the water spec may be quite broad due to adventitious dehydration. Using azeotropic removal of water will work by toluene azeotropic distillation, but you may not recognize the 1H-NMR spectrum of the toluene solution because this dehydration method may produce the dimers, trimers and oligomers from the original organoborate. In my experience, the anhydrides work very well in the Suzuki coupling.
A trial sample of an organoborate must represent what can actually be made at scale. A lab sample will have an analysis of what a skilled chemist can do under optimal conditions. If the customer wants bulk boronic acid matching the qualifying lab sample, then you may be sunk if you cannot reproduce the sample specs in the plant. Been der, done dat.
When submitting an organoboronic acid to a customer for approval, avoid sending the very best material from the R&D lab. They will spec on that sample and expect to see the same thing from a production run.
In the organic synthesis area that I have been witness to, boron showed up in our lives mostly for purposes of aryl coupling chemistry with an aryl halide, an organoboronic ester and a palladium catalyst. The nomenclature gets confusing with boron species. Borax is a borate because of the B-O-B and B-OH bonds. But KBF4, potassium tetrafluoroborate, has a boron-based anion with 4 fluorides. The -ate suffix does not always mean that oxygen is in the formula. Many anionic boron species have a net negative charge because a group like OH or F brought the negative charge to make a tetrahedral boron. Quaternary borates are often used as a weakly coordinating conjugate anions where non-interference by the anion is needed.
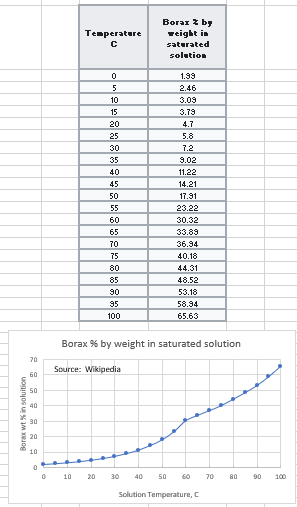
Source: Data table from Wikipedia. Graph by Arnold Ziffel.
Lithium, beryllium and boron are all three transported by meteoric and hydrothermal water flows. When hot, pressurized water penetrates a fault or fracture system, the fluid can interact with whatever rock it is in contact with. Over long periods of time the fluid can corrode the wall minerals of the fault.
In this scenario, water can do several things to the rock. At the atomic level, the outermost layers of a crystal lattice in the mineral may be subject to aqueous hydrolysis making metal hydroxides, MOH, and liberating the anionic X– species from the latticework. Or, the water may just pull unaltered polar species into solution where they can swap ionic partners and either remain in solution or precipitate onto the surface features of the rock. If the hydrothermal fluids are in motion upwards to cooler regions above, precipitation may occur as the fluid naturally cools and depressurizing. Over time, layers of different insoluble material can stack onto the previous layers. This can go on until the void in the rock is filled and can no longer pass fluids through the fractured formation.
Conclusion
Despite the cosmic scarcity of each of the LiBeB elements, these elements may be found on or near the surface of the Earth concentrated as minerals in an ore body. Transport and placement of enriched individual LiBeB ores derives from multiple instances of differential solubility from the source rock to the ore body. Minerals that have more water solubility than adjacent minerals will tend to be transported in aqueous flows. The transported mineral gets concentrated in this way, but so do the minerals left behind. Aqueous flows at the surface can drop out their dissolved minerals as the surface water evaporates. As the surface water concentrates and cools, a sorting process is underway driven by solubility properties.
The original salt ion pair, M+X–, may be extracted into in water and dissociated to produce M+ and X– ions in solution. Other ionic substances like N+ and Y– can switch partners in a double displacement reaction and produce MY and NX. If it turns out that NX has poorer solubility than MY, then the mixture of M+X– and N+Y– will produce a precipitate of NX, leaving much or most of M+Y– in the water. Minerals deposited from evaporation are called evaporites.
Which is more desirable from the manufacturing perspective, the precipitate or the concentrated solution? Not being a mining engineer or an economic geologist, I can only speak as a person from the fine chemical industry. The precipitation of the desired product from a complex mixture seems a bit more desirable in that it is both an isolation and purification process. Often the solids can be washed and dried in a filter dryer and bang, you have your product.
Precipitating out an undesired solid component from a mixture, pure or not, while leaving the desired product in solution with solvent and other side components from the reaction is a bit less desirable. This version is at least another step away. If the product is a solid, perhaps a scheme can be found for precipitating it as well.
Guess what? In response to the Biden’s administration’s efforts to restrict US companies from doing business with 140 Chinese companies, severe export license controls have been put in place. China has responded by banning export of materials critical to the production of semiconductors and other electronic-related articles. This includes gallium, germanium, graphite and antimony- materials required to practice much of our electronics technology. The ban includes diamonds and super-hard synthetic materials (abrasives?). Tungsten, magnesium and aluminum may be next.
The US has previously “restricted advanced semiconductor technology to companies in other countries, though it excluded companies in key allies like Japan, South Korea and the Netherlands that are thought to have adequate export controls of their own” according to Elaine Kurtenbach writing for Manufacturing.Net.
Not helping is Trump’s loud and repeated yammering about import duties. Is he unaware of China’s considerable ability to strike back? Or can’t he be bothered with details like this during his nationalistic diatribes?
Maybe the Biden/Trump Whitehouse is not aware or troubled by the extent to which China has been blessed with most of the world’s supply of many critical elements. The materials subject to Chinese export controls are included in the 50 critical minerals as identified by the US Geological Survey. Key among them are gallium and germanium. Neither of them have been exported to the US by China for a long time. Antimony shipments to the US have plunged as well.
Germanium comes from zinc refining and the US gov’t has a large stockpile. Gallium is a byproduct from bauxite in aluminum production. Antimony is often isolated as a side stream in silver, copper and lead production. Antimony is alloyed at 0.5 to 1.5 % with lead electrodes in lead-acid batteries to harden them. Antimony can be recovered from the lead.
And then there are the rare earth elements (REE). China has the largest deposits and has become unwilling to export unprocessed REEs, instead preferring to sell up the value chain. It is the business savvy way to do it, after all.
Note to readers: The post formerly titled ‘Smartphone Chemistry: An Embarrassment of Riches‘ was poorly titled and has been disappeared. This is an updated version and one titled more appropriately.
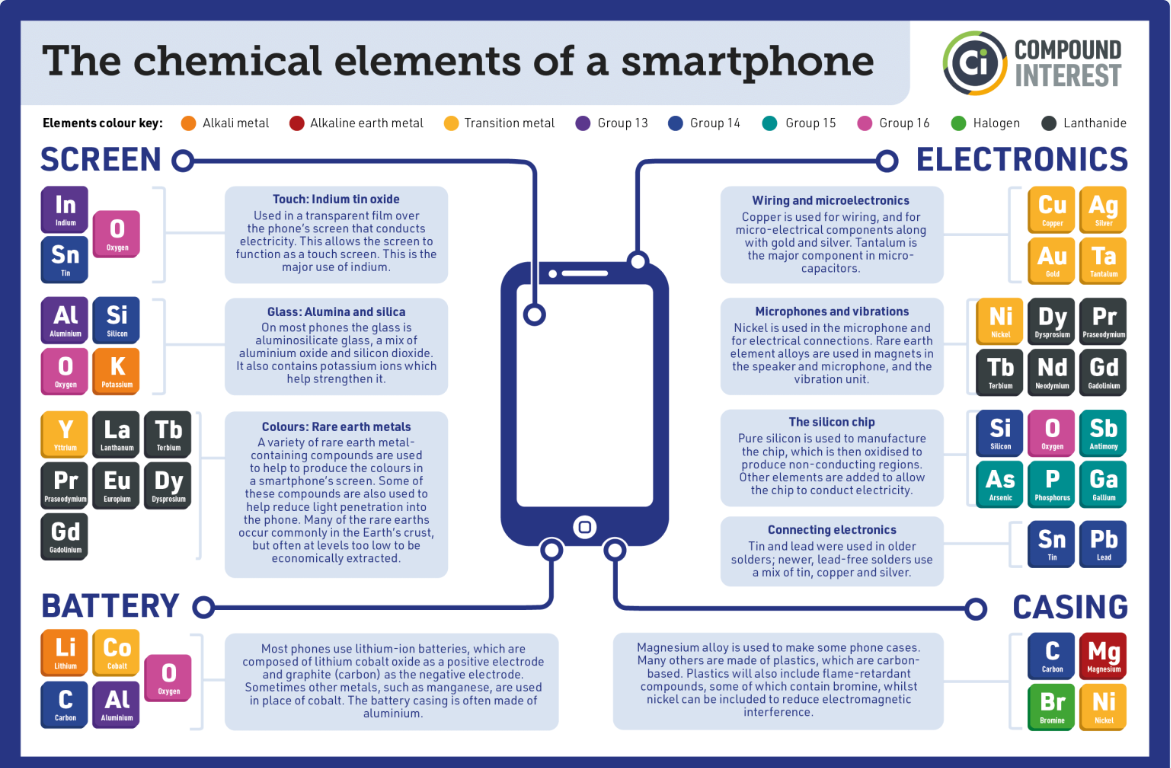
The modern smartphone is made from many different chemical elements, some much more scarce than others. The elements found in a smartphone are distributed around the world, with some countries being more favored by geology than others. With perhaps the exception of silica and silicon, the elements below are found in localized ore bodies that are enriched in particular elements in the form of minerals. ‘Enriched’ is a relative term meaning anywhere between a few 10s of percent to 100 parts per million or less. The word ‘enriched’ also implies another attribute wherein the extraction of the desired element is economically feasible. With the exception of carbon listed below, most of the elements are metals or metalloids. Metalloids are elements that are not entirely metallic but not entirely non-metallic either. Many are found in the p-block of the periodic table.
Source minerals for smartphone manufacture. This public domain image is provided by the USGS.
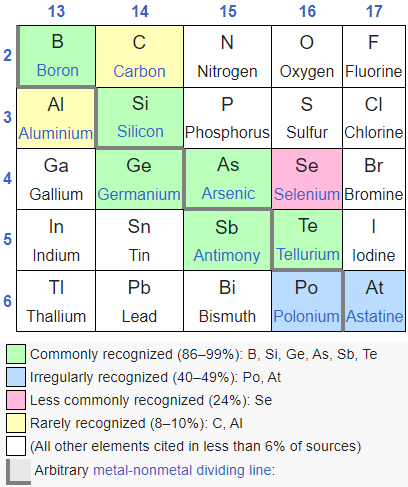
This intermediate chemical nature of the elements we call metalloids may seem a bit dodgy and imprecise, but the term metalloid, while not precisely defined,is thriving out in the big, big world. The metalloids are found in the p-block of the periodic table. Below is a partial chart of p-block elements. Elements polonium and astatine are too rare and too radioactive to be of any practical relevance. It seems as though there is some disagreement as to which elements should be in the set of metalloids.
Source: Wikipedia. The green boxes are the metalloids. They may have some limited ability to conduct electricity or heat, but little of the classic properties of metals like luster, malleability or ductility. The bottom part of the graphic shows that there is some disagreement as to which elements are properly defined as metalloids.
Here is the deal with metalloids. While they may not be used for their physical properties as other metals like in bridge or shipbuilding, their electronic properties provide valuable utility to civilization. By ‘electronic’ I specifically refer to the valence level electrons around the atoms and how they interact.
‘Valence-level’ refers to the outermost layer of atomic or molecular orbitals that are partially filled with electrons. It is at the valence-level where interactions with other atoms and molecules take place. Both the valence-level electrons and empty orbitals are where reaction chemistry occurs.
The black element boxes above represent the Rare Earth Elements (REE) and includes the whole 15 element lanthanide series plus scandium (Sc) and yttrium (Y). Note that Sc and Y are in the same column as lanthanum which is the beginning of the lanthanide series. Generally, elements in the same column share certain chemical properties like in this case the +3 oxidation state, so this is why Sc and Y are considered by some to be in the REE group. The truth is that REE is a woefully antiquated term that just won’t disappear.
“The “rare” in the name “rare earths” has more to do with the difficulty of separating of the individual elements than the scarcity of any of them.” [Wikipedia]
“..,. these elements are neither rare in abundance nor “earths” (an obsolete term for water-insoluble strongly basic oxides of electropositive metals incapable of being smelted into metal using late 18th century technology.” [Wikipedia]
All REEs share the +3 oxidation state, but some of them can have other oxidation states as well. Samarium, europium, thulium and ytterbium can be in the +2 and +3 oxidation states. Cerium, praseodymium, neodymium, terbium and dysprosium all have the +3 and +4 oxidation states. The dissimilarities do not end there. Of the lanthanides, the bookend elements lanthanum and lutetium are often not counted as REEs. The reason is that lanthanum has zero f-block electrons and lutetium has a stable, full f-block of 14 electrons, so neither participates in much f-block chemistry. Lanthanum, [Xe] 5d1 6s2, and lutetium, [Xe] 4f14 5d1 6s2, may be better considered as d-block transition metals.
Snorkeling in Deeper Waters: The notation for the electronic configuration of the elements has a long and an abbreviated form. For heavier elements like lanthanum or lutetium, it is convenient use the abbreviated form showing the nearest noble gas with its orbitals filled- in this case it is xenon, abbreviated [Xe]. Now the orbital levels are filled according to a few rules that look like this for lutetium- [Xe] 4f14 5d1 6s2 . This shows that the f-orbitals are filled with their 14 f-electrons and that the remaining three electrons, 5d1 6s2, are left in partially filled s- and d-orbitals. Filled energy levels (like f14) are very stable and are often quite inert. The three remaining 5d1 6s2electrons are available for chemistry, namely to be removed to form Lutetium (3+).
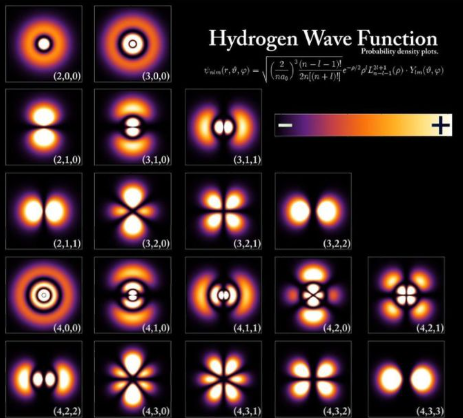
The lowest energy arrangement in which electrons naturally organize themselves under ‘ordinary’ conditions around an atom, molecule or ion is called the ‘ground state’. In the ground state all electrons occupy the lowest energy and oddly shaped regions of space called orbitals, with a maximum of two electrons per orbital. Orbitals are places, not things. There is plenty of information on this quantum chemistry business on the interwebs.
A walk on the wild side
Source: Pinterest. This is a spherical harmonic series of wave functions (orbitals) defining the space that electrons can occupy when in orbit around an atomic nucleus. Each can be occupied by two electrons, but with opposite spins- ‘spin up’ and ‘spin down’.
In the image of atomic orbitals above, each orbital can ‘contain’ one or two electrons. Rather than say ‘contain’, let’s say that the orbitals describe the region of space where the one or two electrons have some probability of being found, depending on their energy. The greater the chance of finding an electron in a particular space, the greater it’s probability density. Note that the orbitals have fuzzy edges. This is because the probability density doesn’t drop abruptly to zero at the edges but rather tapers off. The Uncertainty Principle tells us that it isn’t possible to know both the momentum and the position of a particle simultaneously to high level of accuracy. It turns out that quantum mechanics can’t tell us where an orbital electron is from moment to moment. What it can do is to provide a coherent set of rules for the manner in which electrons are ‘stacked’ in the orbitals as the orbital energy changes.
Alright, we’re back
Scandium and yttrium are d-block transition metals but are sometimes lumped in with REEs because they share the Group IIIB column with lanthanum. The elements cerium through ytterbium do participate in the chemistry of f-block electrons and when REEs are spoken of, there is a good chance that the elements Ce thru Yb are the topic. Is the terminology really as higgledy-piggledy as it appears? Ah, yep.
All of the materials found in electronic devices are there as a result of performance optimization by the manufacturer’s R&D. Many elements are quite expensive, such as indium. The performance uptick from expensive elements must translate into increasing EBITDA. C-suite careers live and die by quarterly and annual EBITDA. Increased performance can be in many forms like longer battery life or increased electronic performance in a chip. Chips require electrical conductors, semiconductors and non-conductors. This is the realm of material science which overlaps with chemistry.
Some of the material science challenges facing smartphone makers might seem a bit arcane. For example, when putting down a layer of material on a chip, will the substrate be wetted by the new layer so that the surfaces contact as desired? An engineering solution may require that a compatibility layer be put down first. Or do the materials have the desired dielectric constants? If you want capacitance in the device, a dielectric layer that is easy and cheap will be required. If you are doing vapor deposition, then the low dielectric material must come from the vapor phase at elevated temperatures. Can it withstand the temperature? Do your semiconductor devices have the desired band gap? What elements affect this? What kind of chemical purity is needed for your CVD or ALD process? Four, five, six or seven nines purity (99.99 % to 99.99999 %)? The more nines of purity specified the more expensive the material and the fewer suppliers there may be.
Companies search all over the periodic table for substances that boost performance and keep Moore’s Law going. All of this must be done in a field full of patent land mines that you don’t want to step on. Invention can lead to big trouble for the unwary.
My, my, my. Rober F. Kennedy Jr. really screwed the pooch with his comments on ethnically targeted COVID-19. Reportedly, he said “there is an argument that (COVID-19) is ethnically targeted”, adding “Covid-19 is targeted to attack Caucasians and Black people. The people who are most immune are Ashkenazi Jews and Chinese …. we don’t know whether it’s deliberately targeted or not.” If this quote is correct, he did not actually say that COVID-19 was ethnically targeted, but rather that “there is an argument …”. It is much like saying “is Bob still beating his wife? I just don’t know …” Whether he endorses the targeting theory or not isn’t clear, but he was willing to trot out this provocative statement to make his point. There was much blowback. Given the racial undertones, it was a large blunder.
RFK Jr. is well known as an advocate for conspiracy theories, some of which are whoppers. The online news magazine Slate has an article that compiles them. I find that his portfolio of mania is exhausting. The thought of pushing back against such seems like a fool’s errand. It reminds of a line in the movie True Grit: “What have you done when you have bested a fool?” What is the point in debating him?
RFK Jr. is a Harvard grad and went the University of Virginia School of Law to get his JD degree. He had a few slip ups early in his career but recovered. He spent most of his career as an environmental lawyer and has fought many laudable battles for environmental justice. Somewhere along the line he went off the rails and landed in the crackpot ferry to conspiracy land. RFK Jr. is a penetrating anti-vaccine voice who can draw large crowds if for no other reason just to see him.
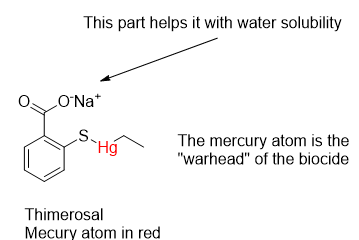
The substance of concern behind much of the anti-vaccine Sturm und Drang is Thimerosal. It is a synthetic organomercurial compound that is effective against bacteria and fungi. Its biocidal properties have been known since the around 1930. Mercurials have been used since the time of the Swiss alchemist Paracelsus (Philippus Aureolus Theophrastus Bombastus von Hohenheim) in the 1500’s. Paracelsus is known for the pronouncement that “only the dose makes the poison.” This remains a fundamental principle of toxicology.
The early mercurial medicaments used by Paracelsus were simple inorganic salts of mercury(II) like mercuric chloride, HgCl2, or mercury(I) like mercurous chloride, Hg2Cl2, also known as the mineral calomel. Mercuric chloride is prepared by treating liquid mercury with sulfuric acid followed by addition of sodium chloride for anion exchange. Mercurous chloride is prepared by heating mercuric chloride with mercury to do the reduction of Hg++ to Hg+.
Thimerosal is sometimes wrongly compared to methylmercury, a known and tragically toxic compound with the formula CH3Hg+X–. The X anion can be chloride, hydroxide or a thiol, depending on the source. It is an easy comparison to make because of the similarity of methyl (CH3) to the ethyl (CH3CH2) hydrocarbon group in Thimerosal, but research has proven it to be a poor comparison. Methylmercury compounds can be produced by aquatic microorganisms in water bodies in the presence of inorganic mercury. The methylation of natural biomolecules is a well-known process.
Like many metals, mercury has an affinity for sulfur, occurring naturally as mercury (II) sulfide, HgS, as deposits of Cinnabar or as a minor constituent with other minerals. It also has an affinity for sulfur-containing amino acids such as methionine, cysteine and homocysteine found in proteins. In the bloodstream mercury binds with proteins like albumin to the extent of 95-99 %. While in the body and exposed to water it decomposes to thiosalicylate and ethylmercury. Ethylmercury cation (CH3CH2Hg+) disperses widely and can cross the blood-brain and placental barriers.
According to Doria, Farina, and Rocha (2015) in Applied Toxicology, a comparison of effects between methylmercury and ethylmercury gave essentially the same outcomes in vitro for cardiovascular, neural and immune cells. Under in vivo conditions, however, there was evidence of different toxicokinetic profiles. Ethylmercury showed a shorter half-life, compartmental distribution and elimination compared to methylmercury. Methylmercury and ethylmercury toxicity profiles show different exposure and toxicity risks.
For many years, Thimerosal was sold as an antiseptic under the name Merthiolate as a tincture (an ethanol solution) by Eli Lilly and Co. Like most households in the 1960’s, we had it in the medicine cabinet or its cousin Mercurochrome. They were used for topical application to burns, cuts and scratches. Thimerosal has been used as a preservative in many health-related preparations such as vaccines, eye drops and contact lens disinfecting solutions. While the CDC has cleared it of doing harm, anti-vaccine mania hit the fan well before COVID-19 and RFK Jr. put his credibility and name recognition behind it.
Thimerosal was first prepared by chemist Morris Kharasch at the University of Maryland in 1927. An interesting technical summary of the substance can be found on Drugbank Online.
Kharasch is known for his pioneering work in free radical chemistry in the 1930’s at the University of Chicago but before that began his work with organomercury chemistry during the 1920’s while at the University of Maryland. His development of Thimerosal was a result of his organomercury work. He is also credited with opening the door to organic free radical chemistry leading to improvements in rubber polymer chemistry and manufacture. His work led to the use of peroxides to reliably induce the so-called anti-Markovnikov addition of a protic acid (HX) to olefins. The presence of trace peroxides was behind the unexpected “reverse” Markovnikov addition of seen in work with the addition of hydrogen bromide to bromopropene.
Kharasch’s early work in organomercury chemistry led to the invention (and patenting) of what became known as Merthiolate (thimerosal). Kharasch later worked as a successful consultant for Eli Lilly, the Du Pont Company, US Rubber, the US Army and others. In many cases these companies were the assignees of the patents.
Little mention is made of Morris Kharasch as a prolific and wide-ranging inventor with, by my count, 117 US patents with him as the inventor. So, why did Kharasch bother to patent Thimerosal? Did he anticipate its biocidal and preservative properties?
Kharasch references make mention of a 1931 patent regarding Thimerosal. That patent is STABILIZED BACTERICIDE AND PROCESS OF STABILIZING IT, US 1862896, appln. filed August 22, 1931, assignee: no party disclosed. The patent claims a process for and claims of water-soluble solution compositions. Numerous additives include antioxidants, alkyl amines, ethanolamine and borax. Claim 19 is telling. It claims the composition of sodium ethyl mercurithiosalicylate (Thimerosal), monoethanolamine, borax as a buffer and enough sodium chloride to make the composition sufficiently isotonic with the body fluids. In this patent the Thimerosal composition itself is not claimed, but as a component of a stabilized water solution. Claim 14 claims a water solution composition of sodium ethyl mercurithiosalicylate and an antioxidant which tends to “inhibit the acquisition” (odd choice of words) of burning properties by the solution. This plus the claim of an isotonic composition strongly suggests anticipated medicinal applications.
STABILIZED ORGANO-MERCUR-SULFUR COMPOUNDS, US 2012820, appln. Feb 17, 1934, assignee: Eli Lilly and Company. Claims a stabilized solution of alkyl mercuric sulfur compounds in water with aliphatic 1,2-diamines. Also claims Ethylenediamine ethylmercurithiosalicylate composition. This is similar to the ‘896 patent but specified ethylenediamines.
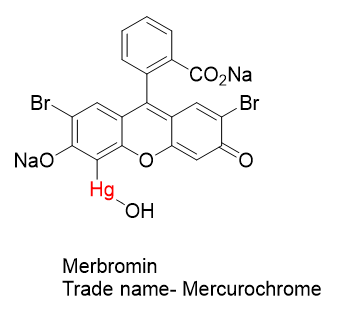
As mentioned above, the biocidal nature of inorganic mercurials had been known for a long time. There was actually limited success in the treatment of syphilis. But they were long known for being very harsh on the patient and grew out of favor when better treatments came along.
The antiseptic properties of Mercurochrome were discovered in 1918 at Johns Hopkins Hospital by urologist Hugh H. Young. Mercurochrome is essentially a dye molecule with an attached mercury warhead. There are three groups on the organic structure that aid in its solubility in water- NaO, CO2Na, and HgOH. Water solubility is often an important attribute in medicinal substances.
Given that antiseptic properties of organomercurials were known, it is perhaps not surprising that an enterprising Ukrainian immigrant with an interest in organomercurials like Morris Kharasch might want to patent his invention.
While cockeyed optimists are working toward a new age of electric vehicles in the glare of an admiring public, I find myself standing off to the side mired in skepticism. What are the long-term consequences of large-scale electrification of transportation?
The industrial revolution as we in the west see it began as early as 1760 and continues through today. Outwardly it bears some resemblance to an expanding foam. A foam consists of a large number of conjoined bubbles, each representing some economic activity in the form of a product or service. A business or product hits the market and commonly grows along a sigmoidal curve. Over time across the world the mass of growing bubbles expand collectively as the population grows and technology advances. Bubbles initiate, grow and sometimes collapse or merge as consolidation and new generations of technology come along and obsolescence takes its toll.
The generation of great wealth often builds from the initiation of a bubble. The invention of the steam engine, the Bessemer process for the production of steel, the introduction of kerosene replacing whale oil, the Haber process for the production of ammonia and explosives, and thousands of other fundamental innovations to the industrial economy played part in the growing the present mass of economic bubbles worldwide.
After years of simmering on the back burner, electric automobile demand has finally taken off with help from Tesla’s electric cars. Today, electric vehicles are part of a bubble that is still in the early days of growth. The early speculators in the field stand the best chance of winning big market share. A major contribution to this development is the recent availability of cheap, energy dense lithium-ion batteries.
Of all of the metals in the periodic table, lithium is the lightest and has the greatest standard Li+/Li reduction potential at -3.045 volts. The large electrode potential and the high specific energy capacity of Lithium (3.86 Ah/gram) makes lithium an ideal anode material. Recall from basic high school electricity that DC power = volts x amps. Higher voltage and/or higher amperage gives higher power (energy per second). Of all the metals, lithium has the highest reduction potential (volts).
Rechargeable lithium batteries have high mass and volume energy density which is a distinct advantage for powering portable devices including vehicles. Progress in the development of lithium-ion batteries was worth a Nobel Prize in 2019 for John B. Goodenough, M. Stanley Whittingham and Akira Yoshino.
All of this happy talk of a lithium-powered rechargeable future should be cause for celebration, right? New deposits of lithium are being discovered and exploited worldwide. But cobalt? Not so much. Alternatives to LiCoO2 batteries are being explored enthusiastically with some emphasis on alternatives to cobalt. But, the clock is ticking. The more infrastructure and sales being built around cobalt-containing batteries, the harder it will become for alternatives to come into use.
One of the consequences of increasing demand for lithium in the energy marketplace is the effect on the price and availability of industrial lithium chemicals. In particular, organolithium products. The chemical industry is already seeing sharp price increases for these materials. For those in the organic chemicals domain like pharmaceuticals and organic specialty chemicals, common alkyllithium products like methyllithium and butyllithium are driven by lithium prices and are already seeing steep price increases.
Is it just background inflation or is burgeoning lithium demand driving it? Both I’d say. Potentially worse is the effect on manufacturers of organolithium products. Will they stay in the organolithium business, at least in the US, or switch to energy-related products? It is my guess that there will always be suppliers for organolithium demand in chemical processing.
A concern with increasing lithium demand has to do with recycling of lithium and perhaps cobalt. Hopefully there are people working on this with an eye to scale up soon. A rechargeable battery contains a dog’s lunch of chemical substances, not all of which may be economically recoverable to specification for reuse. In general, chemical processes can be devised to recover and purify components. But, the costs of achieving the desired specification may price it out of the market. With lithium recovery, in general the lithium in a recovery process must be taken to the point where it is an actual raw material for battery use and meets the specifications. Mines often produce lithium carbonate or lithium hydroxide as their output. Li2CO3 is convenient because it precipitates from aqueous mixtures. It must also be price competitive with “virgin” lithium raw materials as well.
Lithium ranks 33rd in terrestrial abundance and less than that in cosmic abundance. Unlike some other elements like iron, lithium nuclei formed are rapidly destroyed in stars throughout their life cycle. Lithium nuclei are just too delicate to survive stellar interiors. The big bang is thought to have produced a small amount of primordial lithium-7. Most lithium seems to form during spallation reactions when galactic cosmic rays collide with interstellar carbon, nitrogen and oxygen (CNO) nuclei and are split apart from high energy collisions yielding lithium, beryllium and boron- LiBeB. All three elements of LiBeB are cosmically scarce as shown on the chart below.
Lithium is found chiefly in two forms geologically. One is in granite pegmatite formations such as the pyroxene mineral spodumene, or lithium aluminum inosilicate, LiAl(SiO3)2. This lithium mineral is obtained through hard rock mining in a few locations globally, chiefly Australia.
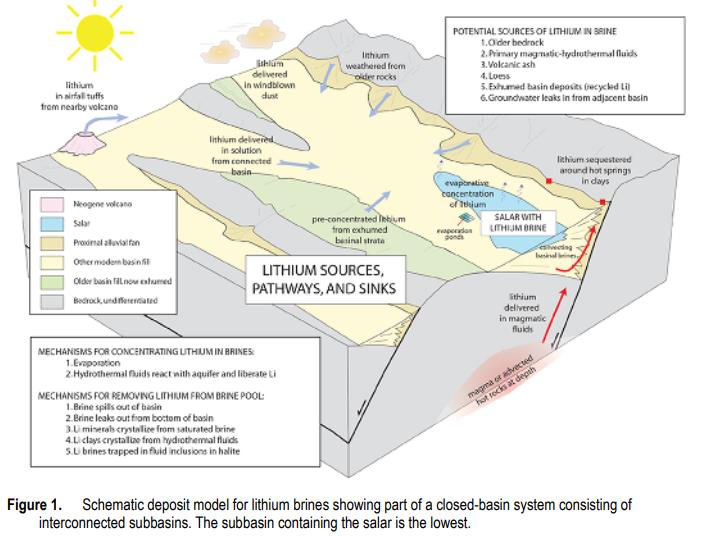
Source: “A Preliminary Deposit Model for Lithium Brines,” Dwight Bradley, LeeAnn Munk, Hillary Jochens, Scott Hynek, and Keith Labay, US Geological Survey, Open-File Report 2013–1006, https://pubs.usgs.gov/of/2013/1006/OF13-1006.pdf
Chemical Definition: Salt; an ionic compound; A salt consists of the positive ion (cation) of a base and the negative ion (anion) of an acid. The word “salt” is a large category of substances, but for maximum confusion it also refers to a specific compound, NaCl or common table salt. In this post the word refers to the category of ionic compounds.
The other source category is lithium-enriched brines. The US Geological Survey has proposed a geological model for brine or salt deposition. According to Bradley, et al.,
“All producing lithium brine deposits share a number of first-order characteristics: (1) arid climate; (2) closed basin containing a laya or salar; (3) tectonically driven subsidence; (4) associated igneous or geothermal activity; (5) suitable lithium source-rocks; 6) one or more adequate aquifers; and (7) sufficient time to concentrate a brine.”
Lithium and other soluble metal species are extracted from underground source rock by hot, high pressure hydrothermal fluids and eventually end up in subsurface, in underwater brine pools or on the surface as a salt lake or a salt flat or salar. These deposits commonly accumulate in isolated locations that have prevented drainage. An excellent summary of salt deposits can be found here.
Critical to any kind of mineral mining is the definition of an economic deposit. The size of an economic deposit varies with the market value of the mineral, meaning that as the value per ton of ore increases, the extent of the economic deposit may increase to include less concentrated ore. If you want to invest in a mine, it is good to understand this. A good opportunity may vanish if the market price of the mineral or metal drops below the profit objectives. Hopefully this happens before investment dollars are spent digging dirt.
Lithium mining seems to be a reasonably safe investment given the anticipated demand growth unless страшный товарищ путины invasion of Ukraine lets the nuclear genie out of the bottle.
Just for fun, there is an old joke about the definition of a mine-
Mine; noun, a hole in the ground with a liar standing at the top.
As interest in lithium batteries continues to ramp upwards, interest in other metals like cobalt used in lithium batteries advances with it. Cobalt has been identified as a particularly problematic metal as the largest single source is in the troubled mineral rich Democratic Republic of Congo. Rightly or not, some are comparing cobalt from Congo to blood diamonds.
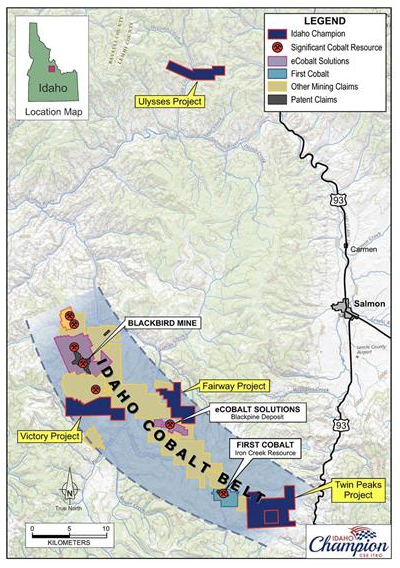
The Idaho Cobalt Belt (ICB) lies on the eastern edge of Idaho along the Montana border running to the northwest for about 60 km. Cobalt was identified in the area in 1900. An abstract in the USGS Publications Warehouse gives a brief technical description of the Idaho Cobalt Belt. The full paper can be downloaded from a publisher’s paywall site. (Why a government agency makes us pay for it’s information work product is beyond my comprehension.)
Another source describes the belt as “originally exhalative, stratiform mineralization within the Proterozoic Yellow-Jacket formation”. The Yellow-Jacket formation is connected with the Yellowjacket mine.
According to EastIdahoNews.com, the Australian firm Jervois Global has suspended the opening of cobalt mining operation in the ICB for an unspecified time. Jervois CEO Bryce Crockersaid the move is “due to continuing low cobalt prices and U.S. inflationary impacts on construction costs.” Over the last 12 months the price of cobalt has dropped from $81,923/T to $34,930/T as of 3/30/23.
The number of cobalt-bearing minerals is too numerous to list here, but a thorough list can be found at minedat.org. Cobalt is rarely found alone, but instead is combined with other metals as a variety of minerals. Important ores of cobalt are Cobaltite, CoAsS, Glaucodot, (Co0.50Fe0.50)AsS, Skuttarudite, CoAs3, and Erythrite, Co3(AsO4)2 . 8H2O. Arsenic is found in 93 cobalt minerals. Other notably abundant elements found with cobalt are sulfur (72 Co minerals), nickel, iron and copper. Other elements associated with cobalt are listed in the minedat.org reference.
The Idaho Cobalt Belt is described as a “strata-bound copper-cobalt district hosted by the Yellowjacket Proterozoic Formation” (1).
According to the Cobalt Institute, there are 5 types of economic geological concentrations of cobalt: Sediment hosted, Hydrothermal and Volcanogenic, Magmatic Sulfides, Laterites and Manganese Nodules, and Cobalt-Rich Crusts.
The crustal abundance of cobalt can be seen in the graph below. From the graph it appears that cobalt, nickel, copper, chromium, zinc, vanadium and manganese have similar crustal abundances.
Cobalt is rarely the sole economic mineral at a mine, at least outside of the Congo. Pyrometallurgy, hydrometallurgy, extraction and electrolytic processing may be used in combination to produce streams of copper, nickel and cobalt. The beneficiation and recovery process of purified cobalt metal or cobalt salt will depend a great deal on the concentration and chemical nature of the elements present.
Cobalt has 2 common oxidation states, 2+ and 3+. According to a USGS reference, the atomic radii of Co2+ and Co3+ are similar to Mg2+, Mn4+, Fe2+ and Fe3+, and Ni2+, meaning that cobalt can substitute for any of these elements in many minerals. The practical consequence of the ability of cations to substitute is a large number of mineral variations. While chemically interesting, this poses complications for ore processing.
Like all metals mining, cobalt must be separated from the ore. Cobalt is not found as the native metal but rather as an ionic complex along with copper and nickel cations. The negative counter-ions are typically silicates and sulfides. Cationic metals ultimately must be reduced to the native metal, but first the desired metal cation must be selectively removed from a dog’s lunch of ionic and covalent species. The desired metal may be removed early in the separation process or after a long series of processing steps to remove other components.
All of the elements and their respective ionic forms have different physical and chemical properties. These differences are exploited in order to separate the elements.
(1) Nold, J.L., Mineralium Deposita, July 1990, Vol 25, Issue 3, p. 163-168. DOI 10.1007/BF00190377
It was announced that a US company will be supplying critical components for Electric Vehicle (EV) batteries to Panasonic. Redwood Materials, Inc., is set to supply EV battery cathode components from its facility in Kansas City. Redwood Materials was founded to close the battery recycle loop by JB Straubel. Straubel was a co-founder and former CTO of Tesla.
A lithium-ion battery doesn’t just rely on lithium. Other substances work together with lithium and the whole composition will vary between manufacturers. The Wikipedia entry for lithium-ion batteries lists the Panasonic cathode material as LiNiCoAlO2. Panasonic works in cooperation with Tesla to supply batteries using Lithium Nickel Cobalt Aluminum Oxide cathode batteries. As alluded to above, Redwood will be supplying cathodes made of recycled battery materials.
The lithium battery electrolyte is almost always contains a lithium salt such as LiPF6, lithium hexafluorophosphate, in a non-aqueous organic carbonate electrolyte like ethylene or propylene carbonate. These two carbonates function as high boiling, polar aprotic dispersants. The substances are cyclic carbonate ester compounds and have a high dielectric constant. The high dielectric constant means that the molecules are polar enough to coordinate Li+ ions to aid in electrolyte mobilization of the Li salt. The electrolyte may also contain a solvent like diethyl carbonate to decrease viscosity and lower the melting point. The PF6 anion is a large, charge diffuse, weakly coordinating anion that helps keep the lithium cation mobilized and loosely bound in the polar aprotic carbonate solution. This anion is inert enough and lends solubility in organic solvents making it useful for many applications. Ammonium salts with PF6 anion are often used as ionic liquids. Weakly coordinating anions are used to allow the corresponding cation to be partially unsolvated and therefore more available for reaction chemistry.
Both in producing power and in recharge, when electrons are being passed around between chemical species and changing oxidation states, it means that chemical changes are occurring. When chemical changes (reactions) are happening, it means that heat is being absorbed or evolved. In the emission of heat, the amount of heat energy per second (power) produced can be large or small. It is critical that the temperature of the battery not exceed the boiling point of the lowest boiling component which may be the carbonate dispersant, as in ethylene carbonate (bp 243 C) or viscosity modifier like diethyl carbonate (bp 126 C). A liquid phase internal to the battery flashing to vapor can overpressure the casing and rupture the battery. A liquid changing into a vapor phase wants to increase its volume by from ~650 to 900 times or beyond. To make matters worse, a chemical reaction generally doubles its rate with every 10 degrees C of temperature rise. Runaway reactions generate runaway heat production.
Lithium batteries have flammable components such as ethylene carbonate (flash point 150 C) and diethyl carbonate (flash point 33 C) that could be discharged and ignited if the battery bursts open, possibly leading to ignition of the surroundings, be it in your pants pocket or in the cargo hold of a passenger aircraft.
In this post I’ll feature a particularly well-done group of videos on precious metals prospecting, milling and smelting. The producer of this content is Jason Gaber at Mount Baker Mining and Metals, MBMM. The website says that Jason is a geophysicist. His company manufactures small-scale industrial grade equipment for the processing of ore. He produces videos that show how things are done in prospecting, mining, and even smelting. His videos give long, lingering views of the milling and smelting processes in operation. I was interested in particular in the process of cupellation, which has always been a bit of a mystery.
Gold ore is dropped into a crusher then pulverized to millimeter-size with a hammer mill. The finely divided ore is then fed onto a shaker table for separation by density with flowing water. The shaker table is a mechanical separation method that allows the isolation of metal fines without chemical processing methods. No cyanide or mercury here. The only waste materials are the pulverized ore tailings.
Editorial comment: To be sure, there is nothing innocent about ore tailings. The large surface area along with the presence of sulfides and water allow air to oxidize the sulfur to strong mineral acid and accelerate the leaching of hazardous metals into streams over the long term. It is very damaging to wildlife and municipalities that draw water from the stream and rivers. Water pollution is a problem all around the American West. Metals are forever.
The smelting videos are interesting for a chemist to watch. Jason uses his knowledge of pyrometallurgy to extract the values and partition impurities away from the target metal. Of course, chemists will recognize this as high temperature inorganic chemistry. Before watching this, I had a poor understanding of the importance of fluxes and slag. Jason quantitatively formulates custom fluxes to fit the problem as he sees it. He uses iron bars for redox processes to change the chemical composition of the melt and give a better partitioning of components.
The goal in smelting is to get a clean separation of the metal value from the ore by partitioning between liquid phases. Lead is often used as a “collector” metal to accumulate reduced metal species as a separate liquid phase on the bottom of the melt. The upper slag phase is a complex mixture of the ore matrix material and contains silicates, aluminates, and a dog’s lunch of other undesirable substances. And. not all metals are miscible or highly soluble in the collector phase, so there is some art in this.
Jason also discusses matte and how to deal with it. Matte is frequently discussed in 19th century works on gold smelting, but this was before atomic theory or sophisticated analytical chemistry. Matte was something to place in a reverberatory furnace and calcine. Sulfides in the matte were converted to oxides and gold residues.
Cupellation is a technique that he uses in the final isolation of gold, silver or PGMs from the collector metal. At the scale of material handling Jason works with, a small cupel and a muffle furnace is all that is necessary for this step. Cupellation for gold isolation was described by Agricola in the 16th century. The lead collector mass selectively oxidizes to the PbO, or litharge, and diffuses into the cupel leaving behind the precious metal. Cupels were formerly made of bone ash or other materials that will not combine with the molten PbO to produce a viscous layer that would prevent seeping of the PbO into the container. This is also how gold was isolated in the old days by the assay office to determine the gold content of ore samples. Today several methods are available to assayers, including x-ray fluorescence.