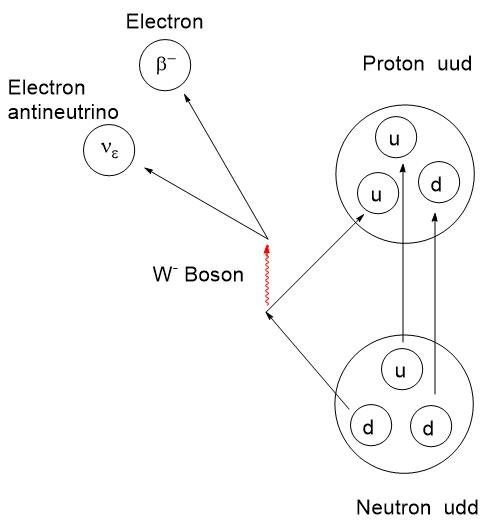
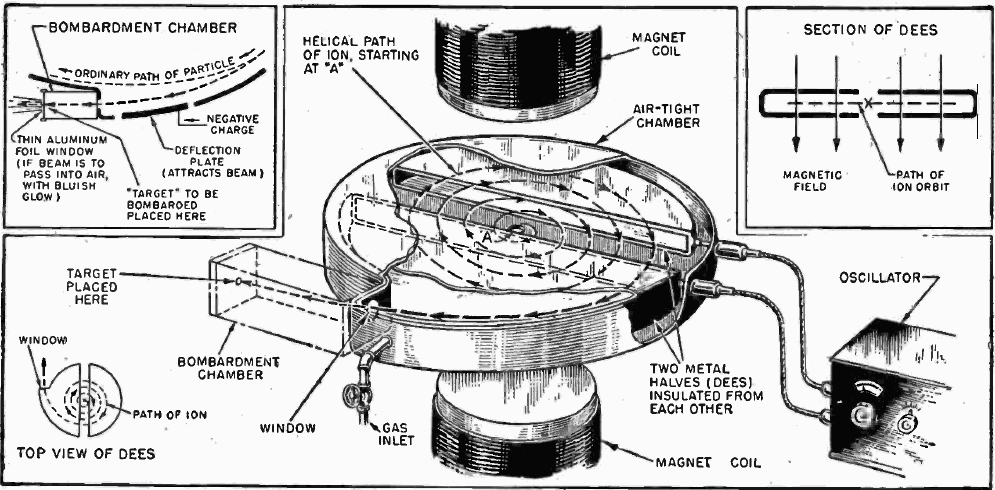
The story of PET, Positron Emission Tomography, has evolved over decades of advancement. To begin, tomography, detectors and computers had to be invented. Separately, positron emission as a medically viable radiation source had to be identified and validated. Positron decay occurs when a neutron deficient nucleus emits a positron and a neutrino to convert a proton to a neutron. This brings the p/n ratio to a more stable state.
A substance for delivering a dose of isotope must be found. In the case of 18Fluorine, it is prepared as an inorganic salt like K18F or elaborated as an organic molecule like 2-deoxy-2-[18F]fluoro-D-glucose.
How did it come about that the 18Fluorine in the position where it is? I’ve not found mention of this in the literature so far. Looking through Chem Abstracts I have noticed there are numerous synthetic pathways leading to fluorine at that position. Could it have been placed there because research found that it was most biochemically similar to glucose? Or was it the more mundane reason that fluorination at position 2 gave the best yields and purity or was the cheapest and easiest to make?
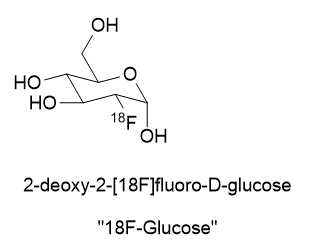
There are recent radioligand compounds that are used as PET (Positron Emission Tomography) diagnostic agents which selectively bind to the prostate specific membrane antigen receptor where they can undergo positron emission revealing the site of prostate cancer cells. 18F-glucose was first synthesized in 1967 in Czechoslovakia at Charles University by Dr. Josef Pacák and was first tried as a radiotracer by Abass Alavi in 1976 at the University of Pennsylvania on volunteers. PET scanning came along later. Cancer cells consume glucose a bit faster than normal cells so the 18F-glucose will tend to accumulate to a slightly greater extent and reveal their position by positron annihilation. This yields two 511 keV x-rays 180o apart and is identified by a ring coincidence detector. A single detection event is discarded.
Today, 18F-glucose is being superseded by many 18F PET preparations that are designed to interact with specific receptors. This interaction is called “conjugation”. In the case of Prostate Cancer there is PSMA, Prostate Specific Membrane Antigen, targeted by Pylarify (piflufolastat F 18) which is designed to bind with fatty acid binding protein 3 (FABP3). I just received a 6 millicurie (222,000 Becquerel) intravenous dose of this positron emitter just today for a PET/CT scan.
Synthetic Strategies Affording 18F-glucose
First, I have to say that the name 18F-glucose is a bit of a misnomer in that it is not glucose nor did it ever even start out as glucose. It is a 2-deoxy-2-18fluoro analog of D-glucose. It originates from D-Mannose whose OH groups were specially protected from side reaction by capping 4 of them with acetyl (Ac) groups and carrying away the hydrogens. The OH at position 2 of the D-Mannose precursor is converted to a triflate (OTf).
In chemical synthesis there is usually more than one possible strategy for getting to a target molecule. In the case of 18F-glucose, whatever pathway we choose must be rapid and efficient owing to the very short half-life of the 18F. The preparation must be done in as few half-lives as possible.
When it comes to a great many sugar derivatives, synthesizing them from scratch is just crazy. They are structurally and stereochemically complex. They have numerous hydroxyl groups in chemically different locations on the molecule and selective modification of one and not another can be quite involved. The world is awash in sugars (e.g., sucrose, starch and cellulose) from natural sources and many varieties are commercially available for developmental use. Better to adapt available sugars for modification than starting from earth, air, fire and water.
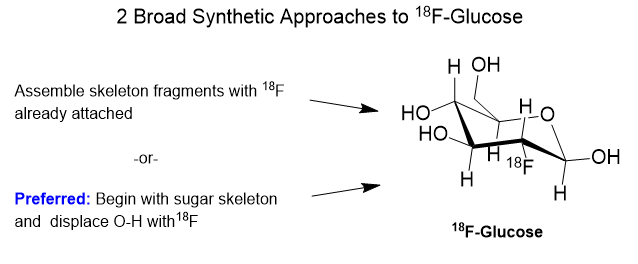
Getting 18F attached to a sugar can go along on one of two basic strategies- electrophilic addition of fluorine or nucleophilic addition. The first is called “electrophilic” addition where electrophile means “electron loving”. In electrophilic addition, the 18F reagent must be electron deficient requiring that the intended carbon skeleton is relatively electron rich. Electron rich means that there are oxygen or nitrogen atoms present with their lone-pair electrons, or pi-bonds present with their off-axis pi-electrons. Equations (a) and (b) below show two examples of electrophilic addition of 18F to a sugar analog.
The fluorinating reagents are (a) 18F enriched F2 and (b) acetyl hypofluorite, [18F]AcOF. Both fluorinating reagents feature fluorine atoms that are electron deficient and therefore electrophilic. Atomic and molecular fluorine are by nature quite electrophilic, but negatively charged fluoride is nucleophilic.
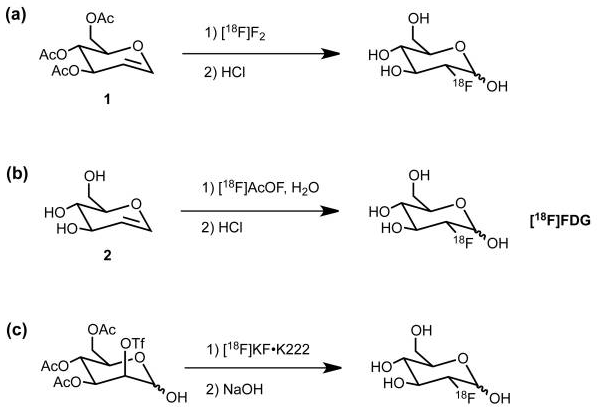
Nucleophilic addition of 18Fluoride is shown in reaction (c) wherein the OTf group is installed specifically to be displaced from the back side by 18F anion. A “nucleophile” is an attacking species that is able to bond directly with a carbon nucleus by virtue of having a lone pair of electrons available for bond making. A nucleophile is frequently negatively charged but can also be neutral in some cases.
The general strategy for the nucleophilic substitution synthesis of 18F-glucose is this: Protect all of the hydroxyl groups of D-Mannopyranose as an acetate except for one which serves as a “leaving group“. This leaving group is called a trifluoromethanesulfonate, or just “triflate“. This triflate is then displaced by 18Fluoride anion by an SN2 substitution. In plain English, 18Fluoride anion forms a C-F bond as the triflate anion is breaking its C-O bond in a process called nucleophilic substitution.
Oh, one more thing. The 18fluoride anion(-) must be made more reactive by keeping the inhibiting potassium cation (K+) in a “cage” so it can lose some of its electrostatic attraction to the negatively charged 18fluoride. Strong electrostatic attraction of K+ to 18F– will impede fluoride’s aptitude for triflate displacement. See below for Kryptofix 222. K+ wrapped in neutral Kryptofix 222 is called a “weakly coordinating ion”.
Ok, so there are some funny things you ought to know about this substitution business on a 6-member ring. Hydrogen atoms are not drawn because it is a pain. First, carbon always wants to have 4 bonds to it and oxygen just two bonds. Second, a 6-carbon ring with all single bonds can be twisted into several shapes or conformations. One of them is favored by virtue of having the least “strain” in it. That would be the “chair” conformation. It looks vaguely like a lawn chair.
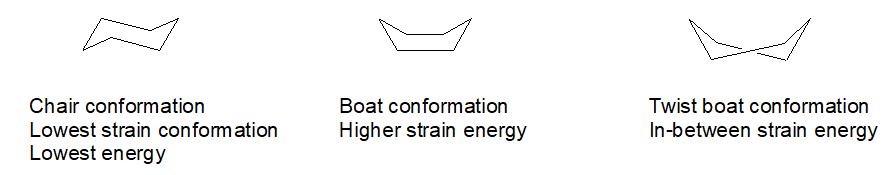
Selective chemical synthesis happens only because some reaction pathways are fast while others are slow. Some possible reaction pathways are so slow that effectively they do not happen.
Making 18F
The 18F isotope does not exist in nature due to its 1.83 hour half-life. It decays by positron and neutrino emission to stable 18O. 18F must be prepared by slamming a suitable precursor nucleus with a nuclear particle like a proton or a deuteron with a cyclotron or linear accelerator. Yes, commercial cyclotrons are available for purchase.
Some Sugar Facts
What helps when thinking about sugars is to detach them from the matter of sweetness. Sugars are far too diverse and important to get hung up on sweetness.
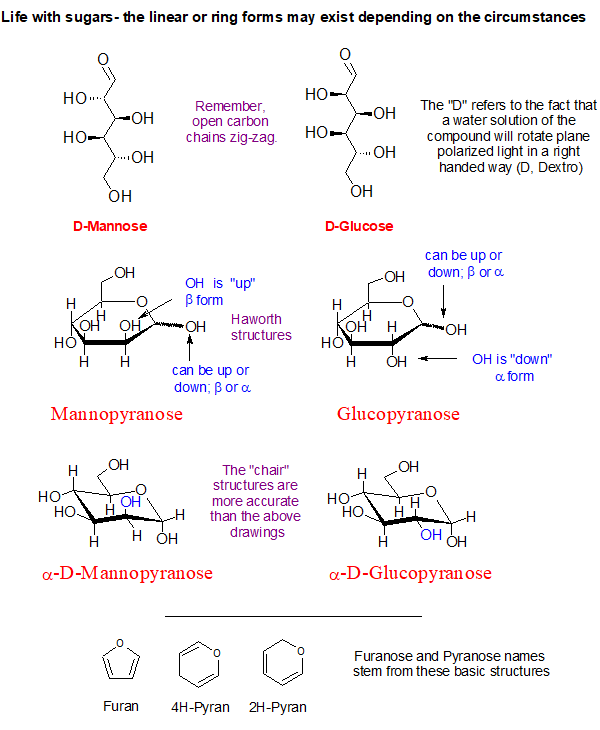
Look at the blue O-H groups on the α-D-Mannopyranose and compare it to the α-D-Glucopyranose shown above. See how they are hanging on the ring? One is directed up and the other is pointing outward and down a bit. This simple inversion in orientation produces the chemical difference between the two sugars making them distinctly different chemical substances.
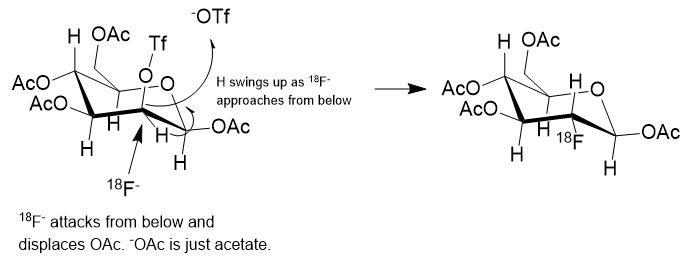
In the reaction scheme above the 18F– is shown displacing the –OTf group from below, establishing a C-18F bond and causing the C-H to flip to the upper side like an inverting umbrella. The scheme is only partially correct. What isn’t shown is the positive counterion to the 18F– anion. The fluoride must be charge balanced by a positive ion which could be just a theoretical bare-naked ion or solvated potassium ion, K+.
In solution, ions or dipolar molecules interact with solvent molecules by Van der Waals forces or stronger dipolar influence. Going down the Group 1 elements on the periodic table from Lithium to Francium, all form 1+ cations, but also the radius of the ion increases. If you think of the ionic radius as being the distance from the nucleus to the distance that a solvent molecule can bump into, the Van der Waals radius, then as we drop down Group 1, a square picometer of “surface” of the ion carries less and less of the cationic charge at any given moment. This means that attractive or repulsive forces with that square picometer diminish as we go down the group, thus lowering the attractive forces. Very often potassium cation is acceptable, but it can be helped along.
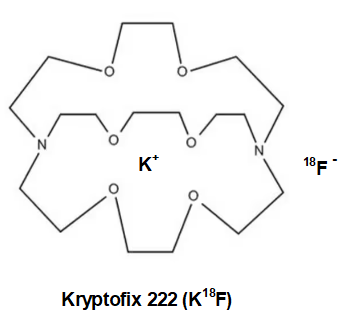
While much of the time K+ is sufficiently non-interfering, but as happens occasionally the fluoride anion tends to bind to the potassium cation a bit too tightly. This can substantially slow the rate of transfer of 18F anion to the carbon of the sugar ring. To get around this, either the potassium must be replaced with another more charge diffuse cation like tetrabutylammonium+ or cesium+, the K+ can be “wrapped” in a protective organic “jacket or shield” that will prevent the K+ and the 18F– ions from getting too close to one another and bound too tightly. We would call the protected K+ a non-interfering or charge diffuse cation.
The cyclic amino polyether “ligand” that is used in this case in Kryptofix [2.2.2]. The single positive charge of the K+ is somewhat spread over the surface of the much larger Kryptofix [2.2.2]-potassium complex and diffuses the positive charge. This has the effect of “separating or loosening” an otherwise tight ion pair (K+F–) in solution. Once detached from potassium, the 18F– ion is able to react much faster to form the 18F-Glucose.
18F-Glucose must be synthesized in a radiopharmacy, also called a nuclear pharmacy, nearby the point of administration to the patient given its very short half-life. The 18F is produced in a commercially available cyclotron or linear accelerator either by proton bombardment of stable but scarce 18O enriched water or by deuteron bombardment of the stable isotope 20Neon.
18F-glucose is a sugar and undergoes metabolic trapping by phosphorylation with hexokinase inside the cell, giving it a phosphate group with a negative charge, inhibiting its transport to outside the cell. This allows the phosphorylated 18F-glucose to accumulate inside the cell, concentrating 18F to release more positron decays from the cell.