I received an email from Academia.org stating that they could turn a research paper which they suggested into a cartoon. Well, what could I do but give it a try?
Cartoon based on a my paper of mine in JOC (long ago) on the facile synthesis of molecules with chiral, enantiomerically pure quaternary carbons. It was a synthetic methodology paper.
Considering that the cartoon has only 4 panels to it, this isn’t so terrible. The title of the paper did have some ordinary vocabulary in it like: the, of, and pure. Isn’t that enough for everyone? Crimony!
In truth this “service” is meant to tickle my funny bone enough to lower my cheapskate defenses, hopefully causing me to subscribe to their service. It didn’t work, this time.
The “facile synthesis machine” is up there with the “Wayback Machine” in terms of wishful thinking.
Summary: The point of this essay is to remind people that, while the works of Charles Darwin and Jean-Baptiste Lamarck were obviously profound in the understanding of many aspects of the biology of life on earth and its adaptation to the environment, their work is very much a product of the mid-19th century. This was prior to the atomic and molecular theories of matter were developed. Since that time the fields of biochemistry and molecular genetics have grown to a high level of sophistication and provided many mechanistic details on how evolution can occur at the level of molecules. With the advancement of biochemistry and molecular genetics, evolution is recognized as a molecular phenomenon using chemical mechanisms not unfamiliar to chemists. It seems likely that if Darwin, Lamarck and others did not make their early contributions to evolutionary theory, biochemists and biologists of the 20th century would certainly have proposed evolution as an inevitable consequence of the mutability of life.
………………..
The frame of reference in this essay is that of an organic chemist’s mechanistic view of the fundamentals of chemical change in biochemistry or molecular biology. Let’s just call it chemistry.
Of the many features of popular science content, one annoyance to the writer stands out: Articles on evolution remain fixated on Charles Darwin’s mid-19th century opus magnum, “On the Origin of the Species“. Darwin’s survey expeditions on the Beagle from 1831 to 1836 as a gentleman companion and naturalist resulted in sharp observations, sample collection, notes, books and years of scholarly lectures.
The question of biological change from evolution dredged up considerable controversy early on, most prominently from the religious communities and lasting to this very day. Much later, after chemistry based on atomic theory was well established, creationists began to sermonize on the statistical problems with the right atoms coming together is the correct order to produce a person. The mantra was that creation implies Creator. Within the context of the Abrahamic religions, the creation of life was clearly stated in religious texts. To assert otherwise was simple heresy. Eventually, the more literate opponents of evolution latched onto the physical principle of entropy.
Entropy is a concept that creationist’s love to unsheathe and swing around. They will say that the 2nd Law of Thermodynamics opens up an apparent contradiction. The crux of their argument is based on equating entropy with ”disorder’. Life itself is comprised of many kinds of highly ordered matter, but the universe is supposed to be getting more disordered. How can this be?
What doesn’t get mentioned is the considerable disorder produced from the life and growth of organisms.
/*BeginEditorial Comment*/
The term “disorder” is the Disneyland word for entropy. It is a highly over-simplified cartoon word meant to describe entropy, which is a thermodynamic state variable. Entropy has the physical units of energy per degree Kelvin per mole. It refers to irreversibly dispersed energy in a system. In my opinion, the word “disorder” is too loosey goosey a definition for even a loose definition.
/*End Editorial Comment*/
A protein molecule doesn’t appear to be “ordered” to the untrained eye. However, a protein molecule has 3 levels of structure in its final construction. First is the specific sequence of amino acids in the protein chain. Second, this protein chain consists of chemical bonds that are free to rotate and chemical bonds that are not as with the peptide bond. This rotational freedom of motion allows a protein chain to rotate about many of its chemical bonds and come to rest in a place where two features may have a reversable mutual attraction. Or, the protein relaxes in a particular configuration that has the least strain.
A third form of protein structure comes from the attraction of individual proteins with another. Proteins often align with another to from large complexes, frequently imbedded in cell walls. These protein structures can contain a channel where ions may pass from interior to exterior of the cell. The channel can be opened or closed in response to external stimulation.
As a protein chain is assembled, it has amino acid features in it that can form hydrogen bonds which allow particular stretches of the chain to reach around and weakly and reversibly connect with itself. An amino acid that has a thiol (or sulfhydryl, -S-H) group can react with another to form a disulfide linkage (-S-S-). The disulfide linkage is a covalent linkage and thus somewhat stable though is subject to reductive or oxidative cleavage.
A length of protein can form a helical secondary structure, a somewhat flattened secondary structure called a beta-pleated sheet, or an unstructured sequence of amino acids.
A few words about entropy, S
One of the ideas frequently cited in creationism is entropy. It is cited because they take evolution as contrary to entropy and the Second Law of Thermodynamics. According to Google, the thermodynamic definition of entropy is given as-
Unavailable Energy: In thermodynamics, entropy quantifies the portion of a system’s thermal energy that cannot be converted into useful work. The more disordered a system, the less energy is available for work.
A more expanded definition is-
Entropy is fundamentally linked to the second law of thermodynamics, which states that the total entropy of an isolated system always tends to increase over time. This means that systems naturally move towards a state of greater disorder and less available energy for work.
The usual interpretation is that the total entropy of the universe is always increasing. This gets interpreted as the world becoming more ‘disorderly’. Unfortunately, the word ‘disorderliness’ can be cognitive bias. The natural meter-scale world we reside in provides many examples of orderliness, which is often just a value judgement by people seeking tidiness.
Creationists often portray abiogenesis and evolutionary change as highly improbable, suggesting the ultra-minuscule chance that the necessary atoms could connect perfectly to create life. If this process was truly random, I might concur. However, the formation of molecules from atoms and the subsequent reactions leading to further changes are not entirely random. Any two atoms or molecules colliding are subject to random motions, true enough. However, what happens during and after a collision and subsequent reaction is far from random. At Earth surface temperatures allowing liquid water, atoms and molecules engage in water compatible reactions, yielding a limited array of possible outcomes and sometimes even a sole outcome. Given certain conditions such as temperature and chemical surroundings, each atom or molecule is restricted to a fairly small number of reaction channels or pathways. Life did not spontaneously arise or evolve from a purely haphazard broth of atoms.
Careless assertions about entropy and the order/disorder of matter can lead to specious conclusions.
The better definition of entropy comes from statistical mechanics. Entropy describes how energy is distributed among the microscopic states of a system. It describes how many ways the system can be arranged at the microscopic level while still appearing the same macroscopically. [From ChatGPT]
Entropy as disorder is perhaps a cul-de-sac rather than the road to understanding.
The Creationist’s respect for the 2nd Law is quaint, but it is more a matter of picking up their opponent’s club and beating them with it in error. When finished, they set it back down and walk away satisfied that they have used science to beat science.
Dipoles- Nature’s Sticky Spots
At the atomic level of matter during a reaction, atoms or molecules may undergo a rearrangement of charge leading to +/- ionic species producing a single pole or a dipole.
Graphic by Arnold Ziffel.
Image by Arnold Ziffel.
When atoms and molecules undergo electronic change the surrounding solvent environment may help or hinder a given transformation. If during the course of a chemical reaction a transient charge is produced, the solvent ‘bag’ enclosing the reacting molecules can promote or hinder the reaction transformation.
My personal policy is to limit the word ‘entropy’ to subjects related to the atomic scale or to heat engines. A loose pile of bricks should rather be described as in disarray.
Molecules and even neutral noble gas atoms can form transient dipoles, causing small, short lived attractive forces between atoms. These are called Van der Waals forces. Graphics by Arnold Ziffel. Graphics by Arnold Ziffel. The ease with which a reaction mechanism proceeds may be subject to solvation effects. Formation of a dipole requires that negative charge is pulled away from positive charge. This takes the application of work against the natural attractive force between positive and negative charges. It takes energy to electronically alter a molecule to produce charge separation to form a dipole. A shell of dipolar solvent molecules around a reacting dipolar molecule can stabilize accumulating polarity in a molecule sufficient to aid the transformation.
Chemical reactions proceed mechanistically in a stepwise manner, and with over 150 years of extensive and peer reviewed chemical research and development, much chemistry has become quite predictable across a wide range of substances. A crucial aspect of modern organic chemistry is the understanding of reaction mechanisms. Biochemists focus on the mechanisms of reaction in aqueous environments, while classical organic chemists commonly avoid water in lab work. However, the principles of physical chemistry support both fields. Indeed, physical chemistry is the cornerstone of the chemical sciences.
On mutation
My greatest hangup about language commonly used to describe evolution is when someone says “The _______ evolved the ability to _______ in order to survive.” True, but In the minds of many this may suggest that a specific genetic change was purposely triggered to achieve the ‘goal’ of enhanced survivability. If a genetic change occurred that improves survivability, it begins randomly. It’s rightly been said that evolution is blind going forward. The DNA of an organism struggling for survival will not automatically give rise to an offspring that have resistance to a given threat. Rather, with each successive daughter cell there is a chance that a beneficial mutation has occurred. But mutations could happen anywhere in the genome. Some mutations may be beneficial, and others may be problematic in terms of survival. There is a chance that the mutation may never be expressed from dormant genes. In order for a mutation to pass forward, it must happen before or during reproduction. Mutations to the parent organism after it has reproduced end right there, beneficial or not.
Radiation-induced mutations to the DNA are more likely to occur during mitosis in cell division when copied DNA strands are being pulled apart and into the daughter cell. The DNA strands are not yet wound with the histones and are more accessible to external influences like radiation or chemical insult.
/* Anecdote: DNA Breakage */
In my second go-around with radiation for prostate cancer in spring of 2024, I learned that the practice now is to deliver approximately the same overall radiation dose as before, but in fewer and larger doses. The idea is to cause breakage in both DNA strands of the double helix rather than just a single strand in the cancer cells. I had 4 sessions of 8 gray in 2024 as opposed to 21 sessions of ~1.5 gray in 2014. I cannot account for why the total dosages are not equal, but there were specific cancerous tissues like the prostate and the seminal vesicles to hit the first time around.
/* End Anecdote */
Today there is a growing understanding that there are actions with the DNA polymer-histone structure that do not involve changes in the genetic sequences. This is called Epigenetics. The total human DNA double helix stretched out is approximately 2 meters in length yet must be contained within a cell. The way that DNA double helix does this is to wrap around a series of individual proteins called histones for compaction into a smaller structure called chromatin. Finally, the chromatin folds into the familiar chromosome structure.
Source: Wikipedia. Public domain image produced by the National Institutes of health.
But is that adaptation by the existing genome composition? Genetic evolution is blind going forward. If a species evolves with a survival advantage of some kind, there can be no “foresight” involved. If it is truly genetic evolution, the end result is because of heritable genetic changes at the level of molecules. If a changing environment causes altered expression of an existing gene in response, say a gene that is otherwise dormant but is suddenly “awakened” by the new environment somehow, then perhaps this is a form of “adaptation within the existing genome” rather than evolution by editing of the genome. This is where epigenetics operates.
Charlie Darwin
The naturalist, geologist and biologist Charles Darwin‘s claim to fame is substantial and well deserved. His book “On the Origin of the Species” is the work that is cited by many but read by few. Over his lifetime he had published considerable work before Origin of the Species. What may be less known is that the notion of evolutionary change wasn’t something that he alone scraped together. Others had previously speculated out loud and in print about changes in species over time. His grandfather, the physician Erasmus Darwin, produced a volume titled Zoonomia that anticipated some of the work of Lamarck which foreshadowed the concept of evolution.
Charles Darwin drawing by Samuel Laurence, 1853. Source: Wikipedia.
In 1831, Charles Darwin embarked on what was originally planned as a two-year voyage of discovery aboard the H.M.S. Beagle, but which ultimately spanned five years. The expedition’s primary goal was exploration, and Darwin, recommended for his scientific interests, joined as a gentleman naturalist with Captain Robert Fitzroy, rather than as a mere specimen collector. Fitzroy, a Vice-Admiral in the Royal Navy and a scientist, led the journey. Darwin, during the voyage, dispatched bones, fossils, seeds, illustrations, and writings back to England, garnering significant interest from geological and natural history circles. Of interest, prior to setting sail Darwin had acquired skills in taxidermy.
After Darwin’s return to England, he spent many years speaking, writing, rewriting and publishing his accounts of the voyage. He is buried in London’s Westminster Abbey just a few meters from the grave of Sir Isaac Newton.
Our acknowledgement of the value of Charles Darwin’s work and methodology is fully legitimate. Darwin’s theory of evolution was a major step change in how we think about biology, speciation and introduced us to natural selection. What is missing from Darwin’s work, however, is the physicochemical mechanism of how evolution works. This is understandable simply because biochemistry was unknown at that time. Inheritance at the molecular level was a mystery until the early-mid 20th century when the molecular biology of the gene began to come together. DNA and RNA had to be isolated and characterized as well as observations made of their x-ray structures. The connection of DNA and RNA polymers to protein formation and composition had to be arduously worked out. Accounts of this are easily found on the internet.
One point of this essay is to argue that while Darwin and others began to coalesce the varied observations of macroscale adaptation and speciation found around the world into a grand theory, the mechanism of evolution lay at the Ångstrom to nanometer scale of the molecule. I find it impossible to deny that if Darwin and others had not come up with evolution at the macroscale, biochemists would have discovered molecular evolution and it would have been used as a basis for the evolution of species.
Sidebar. Rosalind Franklin (25 July, 1920 to 16 April, 1958)
Much acrimony has been made over the alleged snubbing of physical chemist Rosalind Franklin in the selection of 1962 Nobel Prize winners in Physiology or Medicine for the discovery of the structure of DNA. A recent paper in the 25 April, 2023, issue of Nature brings together some little-known details of the cold-shoulder given Franklin as a co-discoverer of the double helix structure of DNA.
The disqualifying event for the 1962 Nobel Prize occurred in 1953 with Franklin’s death. At the time, posthumous awards were accepted only if the death occurred between nomination and the award date which was not the case for Franklin.
The story of the discovery of the double helix structure of DNA involves 4 central characters: James Watson and Francis Crick from the University of Cambridge, UK; Rosalind Franklin and Maurice Wilkins at King’s College, London.
Before any of this started, the involvement of deoxyribonucleic acids in heredity had been suggested by Oswald Avery in 1944 (below). Watson and Crick, Franklin and Wilkins did not discover DNA. They did, however, use x-ray diffraction of crystalline DNA fibers and model building to deduce the chemical structure of B DNA.
At King’s College at the time of this story, the biophysics group was led by John Randall whose deputy was the New Zealand-born biophysicist Maurice Wilkins. In 1951 Franklin joined the Department on a 3-year fellowship having come from Paris where she used x-ray diffraction to study the structure of coal. By this time Wilkins had been working on DNA since 1948. A personality clash arose between the more assertive Franklin and the less confrontational Wilkins, so Randall divided certain DNA samples between them. Franklin received a calf’s thymus-derived sample of the highly purified material from the Swiss chemist Rudolf Signer – Wikipedia at the University of Bern and Wilkins received a “poorer sample” from Chargaff at Columbia University in NYC.
Sidebar to the sidebar.
Chargaff had become interested in DNA after Oswald Avery at the Rockefeller Institute published a paper in 1944 concluding “The evidence presented supports the belief that a nucleic acid of the desoxyribose type is the fundamental unit of the transforming principle of Pneumococcus Type III“. Avery and colleagues had developed the first immune serum for a strain of pneumococcus from the blood of horses. Along with colleague Michael Heidelberger they found that polysaccharides associated with this strain of pneumococcus could be isolated from water-soluble spherical capsules around the cocci and are antigens, which later led Heidelberger to discover that antibodies are proteins. These are now fundamental facts in molecular biology and immunology.
Back to the double helix
Wilkins had earlier discovered that there were 2 forms of the DNA in solution- the crystalline A form and the paracrystallineB form. Franklin took the A form and Wilkins the B form. Franklin discovered that the A form will convert to the B form in higher humidity and revert back to the A form in lower humidity.
Photo 51. The x-ray diffraction pattern of the “B” form of DNA, taken by Raymond Gosling while working under Wilkins. Source: Wikipedia
Unfortunately for Franklin with the A form, Wilkins’ B form is what is found in the cell.
From a biochemical standpoint, mutation of DNA sequences makes chemical sense and today is routinely observed in DNA assays. An increasing number of diseases or characteristics are linked to distinct mutations in DNA. Deoxyribonucleic (or desoxyribonucleic) acid (DNA) is a chemical substance that, like all chemicals, is susceptible to its chemical environment and whatever particular substances happen to be nearby or to ultraviolet or ionizing radiation or to highly reactive chemical species like free radicals.
Left to right: A, B and Z DNA structures. Image from Wikipedia. Note the difference between Franklin’s A-DNA vs Wilkin’s B-DNA. They differ by the extent of hydration.
Biochemical Evolution
Charles Darwin is renowned for his well-articulated, evidence-based argument on the evolution of species by natural selection. He courageously introduced a detailed new theory within the conservative British scientific community. His ideas fascinated many leading naturalists of the time, who adopted and furthered the theory. Truly, it marked a considerable progression for that period.
“But but but, it’s just a theory!” This is a common objection by creationists and religious zealots thinking they have found the weak underbelly of evolution. They claim it is “just” a theory as though a theory was merely a fanciful excursion of the imagination where all opinions are of equal validity.
A theory is an overarching explanation or model subject to improvement over time with which arguments are made in support of or against a core concept. This core concept is initially built on a pedestal of clay. As better analysis and experimental data come in, the pedestal is strengthened or weakened. Furthermore, the theory may be unequally affected across its breadth with some aspects perhaps tossed out and others supported. Theories themselves evolve and strengthen with evidence. Scientists are naturally anxious to contribute to sorting out the truth of a theory.
Another objection to evolution is the previously stated notion that “creation implies the existence of a creator.” A common argument is that a watch is such an unlikely collection of highly refined components that there must be a watchmaker behind it. The human hand or eye are the anatomical examples often cited. There is a bit of vocabulary that muddles these arguments. The use of the word “creation” presupposes that the universe is something that was assembled by a creator. If you see the world as something that had to have been created, then the idea of evolution may be difficult to swallow.
The question of life on Earth has two important aspects to it. One is the evolution or change that species undergo over time. The other is the initiation or abiogenesis of life from non-living matter. Of the two areas, the evolution is the most developed concept.
Biochemists and molecular biologists have taken Darwin’s evolution from a mid-19th century macroscopic theory supported by the fossil record, geological observations and the gross anatomy of animal species from around the world to the submicroscopic machinations of molecules. This has been a gigantic leap forward in understanding not just in the chemistry of all life but also the evolutionary physicochemical mechanisms of life. Life is one of the things that chemicals can do given opportunity and time.
Christian and other churches reacted negatively to evolution in Darwin’s time as most do today. Darwin and many geologists concluded that the Earth was far, far older than did scholars withing the church. So, what is this about? Are church leaders skeptical or just stubborn? Is this even a good question?
Types of thinking
I would offer that people can be spread between two bookends in regard to thinking: Devotional thinkers and analytical thinkers. Devotional thinkers have a core doctrine supporting their beliefs and think and behave in a way that their belief guides them. Devotional thinkers study their doctrines in an effort to be in better alignment with it. It is not uncommon for devotional thinkers to limit their exposure to things not aligned with their devotion. Devotional thinkers are sometimes labeled faithful. Their goal is to study supernatural doctrine and align one’s personal behavior.
Analytical thinkers will naturally adopt a baseline worldview that comports with their education, observations and logical sensibilities. But when presented with new data or just a compelling idea, analytical thinkers may be persuaded to open new vistas in their thinking or at least set the idea aside as new thinking under consideration. Analytical thinkers are sometimes labeled as skeptics.
It is impractical to approach each new circumstance one encounters ab initio. In order to explain how an airplane flies it is not presently necessary to first independently derive Newton’s laws of gravity and Prandtl’s fluid dynamics so as to set the stage for lift and drag forces. Everyone has a practical baseline picture of the world that serves as a conceptual starting point for some kind of conclusions on reality. The discipline of science is highly vertical with old knowledge built upon or revised by new knowledge. The requirement for accuracy is practiced by the investigator and checked upon by peer reviewers. Nobody wants to be that scientist who has published a paper with faulty science requiring a retraction in the Retracta Acta.
To be skeptical of the evolution of the species is on one hand to require supporting evidence and compelling arguments. On the other hand, many people dismiss evolution altogether as being contrary to their faith-based notions while posturing as an “evolution skeptic.” However, when physical evidence or collected data are thrown on the table for all to examine and when that evidence is part of a trail of evidence logically or mechanistically interconnected, then to dismiss the logic or measurements is to go beyond skepticism. Apologists would claim that they are keeping their faith against adverse influence or even resisting evil. But standing against evidence could really be considered simple stubbornness for fear of perceived divine consequences or discomfort.
DNA and RNA are polymeric substances comprised of four major subunits. Three of the subunits are shared by both DNA and RNA and the fourth is a different component characteristic to RNA. This determination took some time to arrive at the correct structures and the chemical mechanisms.
Graphic by Arnold Ziffel.
Chemicals interact by particular mechanisms depending on what is present and physical conditions like temperature, pressure or interfering substances. These mechanisms are a built-in, reliable feature of matter in our universe. When multiple mechanisms are possible, the fastest one tends to prevail, channeling matter down that pathway. The fastest channel will have to lowest energy barrier to cross. However, if the reverse mechanism is possible then a balance will be struck between two reservoirs of substances. This is the basis for thermodynamic equilibrium. The reaction direction with the lower energy barrier will be faster, and if the reverse direction isn’t possible, the mechanism will preferentially populate the direction with the lower energy barrier. We would say that the reaction is under kinetic control. If the reaction can go both ways, then a balance will be struck producing substances on both sides of the energy barrier producing thermodynamic control.
One argument offered by Creationists is that the probability of all the atoms in a human coming together to form that human is 1 in 10stupid large. In other words, they say, highly improbable within the age of the universe. And if that was how it works, then I’d agree. But it is definitely not how chemistry and evolution work.
Evolution happens by a biochemical ensemble of mechanisms in solvent water constrained by the boundaries of chemistry and physics and specifically to what is possible in aqueous media. Liquid water is necessary rather than solid or crystalline phase water because for bio- or any chemistry to operate, molecules have to diffuse around and collide in order to react. Biochemistry and therefore evolution occurs at temperatures between roughly -10 oC and 45 oC, plus or minus a bit and at midrange pH levels.
Life is substantially based on carbon because carbon forms stable chemical bonds with nitrogen, oxygen, sulfur, hydrogen and especially with itself. Phosphorus appears as phosphate. Carbon can form chains of indefinite length and 3, 4, 5, 6, 7 and 8-membered rings or larger with 5 and 6 being the most common ring sizes. Tens of millions of different chemicals structures are possible within this group of elements. Nature is crammed with ring systems in natural products.
A line drawing and 3D rendering of Taxol or Paclitaxel. Silicon does not do this. Image: Wikipedia.
Biochemistry is not based on silicon, even though silicon has certain chemical similarities to carbon. Silicon does not easily form chains greater than 2 silicon atoms in length and it has a strong affinity to oxygen. This affinity is very much thermodynamic in nature and is difficult to overcome chemically at biological temperatures and pH. Silicon-nitrogen bonds are hydrolytically unstable at low to moderate pH. All of this adds up to poor utility for silicon in biomolecules.
Each of these carbon-nitrogen, carbon-oxygen, carbon-phosphate, carbon-sulfur, carbon-hydrogen and carbon-carbon bond combinations as well as the various combinations of N, O, P, S, H atoms have their own variations as well. Other atoms like iron, calcium, sodium, potassium, magnesium, chromium, selenium, iodine, and a few others serve purposes other than for molecular skeletons generally.
The point of citing all of these combinations of atoms is to emphasize that each has unique chemical properties and unique reactivities. The slapdash Creationist assertion that evolution merely brings atoms together to form an organism and no consideration of reactivity is mentioned. While molecules in solution are more or less randomly banging around, their entry points for successful chemical interactions are far from completely random. In fact, a given molecule will react only in a few ways depending on what it collides with. A complex molecule like glucose has several reactive sites, but it still has a limited menu of reaction types available at physiological conditions.
Molecules are tiny objects that can have even smaller features where changes can happen. These features can only do certain kinds of chemical reactions. They are called ‘functional groups’ and they are limited to a limited set of reaction mechanisms, often only one mechanism.
Think of each of these limited types of reactions as a channel. Overall, biochemical transformations happen through these particular channels. There may be numerous channels possible on a molecule affording diverse reactive outcomes. Even among the possible transformations, a few channels will react faster and thus dominate. The point is that there are not an infinite number of ways that molecules can exhibit reactivity. This means that evolutionary change through biochemical mechanisms does not have an infinite set of likely chemical pathways. There can be many, to be sure. But the entire ensemble of biochemical mechanisms operating in an organism do not have to change to allow a given evolutionary change.
Evolutionary changes can occur in very subtle ways. A biochemical modification may result from a misreading of the normal genetic code or from some other off-normal situation, but this is not an evolutionary change. A genetic change can result from an alteration in the sequence of the genetic code itself. A change in the sequence of the DNA may lead to a heritable mutation if the change is survivable. A mutation in an unused stretch of DNA may occur and lead to no effect. If the genetic change is fatal to the new cell, cell death can occur, and the mutation will not be passed forward.
Cosmic ray showers. Image NASA. High energy cosmic rays impinge on the upper atmosphere and collide with air molecules, causing nuclear reactions that result in showers of nuclear particles like muons. Most of us do not realize that we have muons in our lives, but there we are.
Energetic cosmic radiation from outer space and the sun is constantly showering the Earth’s upper atmosphere. When a particle or a photon from space impacts a person, it will penetrate to some depth, dumping kinetic energy into tissues that can break chemical bonds to form ion pairs or radical species. Ion pairs can reconnect as before or with other species to form new substances. Radicals are neutral atoms or molecules where electrons in the form of lone pairs or covalent bonds are evenly split into 2 radical species where each is neutral but have unfilled octets. Radicals tend quench themselves by popping off a hydrogen radical from a nearby molecule or by colliding with the radical that was originally separated.
Radiation exposure of living tissue or other material objects produces ‘stochastic’ damage because the kinetic energy of the radiation particle or photon far exceeds the energy needed to cause bond breakage or general scrambling of biomolecules. Stochastic radiation damage is fairly unselective so, as the radiation passes through materials, the energetic particle dumps some or all of its energy into the material directly along its path of movement.
One measure of the potency of a given particle or photon of radiation is the number of ion pairs produced per inch or centimeter. The three major types of radiation are alpha, beta and gamma. Alpha particles produce the most ion pairs because of its high kinetic energy so it dumps its kinetic energy along a very short distance. Beta particles can travel a bit longer distance (several inches) but require less shielding than gamma rays. While gamma rays are quite penetrating, their ion pair production is very low.
Any given human exposure to radiation can result in no observable effect or tissue damage. Not every exposure to radiation will result in cancer. Because radiation damage to tissues is stochastic, a survivable mutation of DNA is random.
Enzymes are proteins that act as catalysts or enablers of chemical transformations. These protein enzymes have places on them that are clefts, ridges and valleys along their exterior where other molecules or coenzymes can collide and interact called an active site. It is possible for the enzyme to be active continuously and catalyze transformations when the right substrate molecule jostles along. It is also possible for the enzyme to require ‘activation’ in order to function. One means of activation comes from phosphorylation of the substrate to be acted upon, phosphorylation of the enzyme itself, while yet another is where an external molecule binds to a particular spot, resulting in an alteration in the overall shape of the enzyme. This alteration in the enzyme’s shape can open the active site of the enzyme and allow the intimate contact necessary for a particular molecule to diffuse in, bind and undergo a catalyzed transformation. In this way, an enzyme can be deactivated as well.
Much new drug discovery and design is based on toggling an enzyme “off or on” with a suitable substrate. The substrate can be constructed so as to be highly specific, to be detachable or to sacrifice itself by covalently connecting to the enzyme and prevent it from further functioning. This last category is sometimes referred to as a suicide substrate inhibitor. Penicillin is an example of a suicide inhibitor that covalently combines with an enzyme, shutting it down permanently. Penicillin and its many analogs have a strained 4-membered ring in them called a beta lactam ring that can relieve the strain by ring opening to a straight chain by connecting to a feature on the enzyme. This is irreversible though bacteria have developed the ability of reacting with penicillin to eliminate its ability deactivate a target enzyme.
Enzymes can be very sensitive to a change of one amino acid which could lead to little change or it could cause the enzyme to operate a few percent faster or slower. Or it could change the reaction rate or specificity by a great deal. It might even allow different substrates to be acted upon by the enzyme. Let’s say that this change in the operating rate of the mutated enzyme causes a chain of successive biochemical reactions to operate faster, say by increasing the efficiency in the use of energy. If this results in the survival rate of the organism increasing by a bit, it may impart a survival advantage. If the alteration of the enzyme reduces the survival rate, then the organism may continue to survive or not.
But hold on. Evolution is blind going forward. Not all changes register as an advantage or even show an effect. If the mutation happens after reproduction, then it does not get inherited by succeeding generations. If the mutation is fatal, then the cell dies and the genetic change halts. If the process of evolution is so iffy, how does anything happen?
We should reflect on how fast chemical reactions can happen. At room temperature, water is undergoing collisions at a rate of ~1010 per second. Now imagine 1 mole of water, 18 grams, all molecules undergoing ~1010 collisions per second. One mole of water contains 6.02 x 1023 water molecules. Simple mindedly, that adds up to 6.02 x 1033 collisions per second in those 18 grams, or 1 thirsty swig of water. But that’s not all. These water molecules are also vibrating, rotating and translating at perhaps 1012 vibrations per second. In general, each collision will carry a particular probability of a bond breaking or bond forming event. Water is a boring example, but we can see that a reactive biomolecule is also undergoing a very large number of collisions per second, each with a certain chance of participating in a reaction. Even though a given reaction may be of low probability per collision, a great many collisions raise the odds of fruitful interaction.
Rapid molecular collisions in combination with a limited range of reaction channels means that the molecules will sort themselves out by way of finding the lowest energy-barrier, fastest reaction channel to follow. This is far from completely random. The fastest reaction channel can consume its inputs the fastest and the product from this fast channel will predominate.
Why would there be DNA, protein or other biomolecules that are fragile enough to suffer mutation in the first place? Why hasn’t DNA evolved into a sturdier structure free of mistranslation, mutation and other errors in its functions? There are certainly substances that are more robust than DNA or RNA like hydrocarbon polymers, silicates, urethanes, urea linkers and other polymers that are much more stable to chemical insult. The DNA double helix is, after all, held together by low energy hydrogen bonds.
A key requirement of life as we know it is that something has to prompt DNA to unravel and split strands of deoxyribonucleic acid chains apart. In order to unravel, the structure holding it safely in the double helix form must be capable of assembling and coming apart when prompted. The nucleic acid structure along each chain of the double helix have phosphate linkages. Because phosphoric acid is a weak mineral acid it can lose one, two or three acid protons (mono- di- or tribasic) under physiological conditions.
The phosphate linkage in DNA works very well for life. It allows free rotation about the linkage and is quite polar for good compatibility with water. Phosphate can form bonds between themselves: 1, 2 or3 phosphates can link, leading to short chains of phosphate anhydrides. Because phosphate is relatively stable under physiological conditions yet is able to function, it is nearly ideal for its purpose. Phosphate is phosphorus (V) with 4 oxygens bonded in a tetrahedral fashion. Three of the oxygen atoms have single P-O bonds with one P=O double bond. When connected as anhydrides, mono-, di- and triphosphate anhydrides may form. The linking oxygen atom connecting the two end phosphates can be displaced and added to another substrate. This is called phosphorylation and is critical in biochemistry.
Abiogenesis
Abiogenesis is the big puzzle at this point in history. Evolution is not the same as abiogenesis. How did life begin? As we look around at the Earth today, we see an overprinting of billions of years of planetary, geologic, oceanic, and atmospheric transformations. The present world at the surface is nowhere near that world at the time when life began to flicker into existence. One of the primary differences is the chemistry in play. Before oxygen began to accumulate in the atmosphere, many elements may have been exposed at a reduced oxidation state, that is to say electron rich. The chemistry of an atom greatly depends on the state of its valence electrons. So much so that the atom loses its identity when charged or in molecular form. For instance, +H (protium or hydrogen cation) is chemically different from –H (hydride ion) is different from 0H (atomic hydrogen)and all are different from molecular H2. Referring to +H as hydrogen is incorrect. It is properly referred to as “hydrogen ion” or “protium ion”. The ions are distinct chemical species. This range of possibilities in the state of reactive atoms (other than the noble gases) somewhat complicates the chemistry of prebiotic Earth.
At this point I’ll refer the reader to the InterWebs for deeper insight into abiogenesis.
In a YouTube interview recently, Roger Penrose commented that one of his beefs with quantum mechanics (QM) was that it depends on consciousness collapsing a wave function. He said much more, but this struck me deeply. I have been struggling for years trying to verbalize my own brain-stem level suspicions. (To be clear, a Venn diagram with Penrose and myself overlap only insofar as we are both English speaking bipedal mammals.)
He also commented that by “wrong” he means “incomplete” and recalled that both Einstein and Schrodinger agreed on this. He noted that “incomplete” constitutes “wrong”. In another interview he comments that Schrodinger’s Cat was meant to illustrate that the superposition of a cat being both dead and alive was a problem that Schrodinger recognized. A cat cannot really be dead and alive simultaneously.
He said that while QM was about more than the evolution of a quantum state, it is also about measurement, but the measurement problem violates the quantum equation. QM gives the probability of a given state which is a superposition of probabilities.
QM is not my specialty, however, I have coursework in it like all chemists have had. In grad school I had quantum chemistry, again like all chem grad students have had, but it nearly did me in. Not having had a semester of diff eq, I was at a distinct disadvantage. Grad school QM goes well beyond the particle in a 1-dimensional box. The course consisted of mathematical derivations of the theory, but not much about the meaning in English. We were supposed to see the equations in their abstract purity and extrapolate to some kind of comfort level with notions our brains could grasp. It was based on the Copenhagen Interpretation of QM. Philosophically, the notion that the wave function would collapse on inspection by a brain was an idea that even today I cannot get past.
Penrose had a similar beef except that he is Roger Penrose and I’m some lesser ape gawping in from up the holler. Still, though I’m doomed to go to my grave with only very rudimentary understanding of QM, the concept of probability density all by itself as well as the spherical harmonics defining atomic orbitals has been a major benefit for my thinking. And for that I’m grateful.
Below is a cut & paste copy of text from Wikipedia outlining Penrose’s ideas-
Penrose’s idea is a type of objective collapse theory. For these theories, the wavefunction is a physical wave, which experiences wave function collapse as a physical process, with observers not having any special role. Penrose theorises that the wave function cannot be sustained in superposition beyond a certain energy difference between the quantum states. He gives an approximate value for this difference: a Planck mass worth of matter, which he calls the “‘one-graviton’ level”.[1] He then hypothesizes that this energy difference causes the wave function to collapse to a single state, with a probability based on its amplitude in the original wave function, a procedure derived from standard quantum mechanics. Penrose’s “‘one-graviton’ level” criterion forms the basis of his prediction, providing an objective criterion for wave function collapse.[1] Despite the difficulties of specifying this in a rigorous way, he proposes that the basis states into which the collapse takes place are mathematically described by the stationary solutions of the Schrödinger–Newton equation.[4][5] Recent theoretical work indicates an increasingly deep inter-relation between quantum mechanics and gravitation.[6][7]Wikipedia.
I just donated to Wikipedia so I don’t feel too bad about this cut & paste. Please donate to Wikipedia. We want to avoid a paywall being put up in front of it.
Today I have a slightly different demographic of readers of this blog than in the past, so I’ve been dredging up old posts into the light of day. This is a renamed post from September 3, 2011. I’ve changed some wording to be a bit more mellifluous if that’s even possible.
==========
I’ve had this notion (a conceit, really) that as someone from both academia and industry, I should reach out to my colleagues in academia in order to bring some awareness of how chemistry is conducted off-campus. After many, many conversations, an accumulating pile of work in local ACS section activities, and visits to schools, what I’ve found is not what I expected. I expected a bit more academic curiosity about how large-scale chemical manufacturing and commerce works and perhaps what life is like at a chemical plant. I’d guessed that my academic associates might be intrigued by the marvels of the global chemical manufacturing complex and product process development. Many academics would rather not get all grubby with filthy lucre. Not surprisingly, though, they already have enough to stay on top of.
What I’ve found is more along the lines of polite disinterest. I’ve sensed this all along, but I’d been trying to sustain the hope that if only I could use the right words, I might elicit some interest in how manufacturing works- that I could strike some kind of spark. But what I’ve found is just how insular the magisterium of academia really is. The walls of the fortress are very thick. I’m on a reductionist jsg right now so I’ll declare that chemistry curricula is firmly in place on the three pillars of chemistry- theory, synthesis, and analysis. In truth, textbooks often set the structure of courses. A four-year ACS certified chemistry curriculum spares only a tiny bit of room for applied science. I certainly cannot begrudge departments for structuring around that format. Professors who can include much outside the usual range of academic chemistry seem scarce.
It could easily be argued that the other magisteria of industry and government are the same way. Well, except for one niggling detail. Academia supplies educated people to the other great domains comprising society. We seem to be left with the standard academic image of what a chemical scientist should look like going deeply into the next 50 years. Professors are scholars and they produce what they best understand- more scholars in their own image. This is only natural. I’ve done a bit of it myself.
Here is my sweeping claim (imagine waving hands overhead)- on a number’s basis, chemists apparently aren’t that aware of industrial chemical synthesis as they come out of a BA/BS program. That is my conclusion based on interviewing many fresh chemistry graduates. I’ve interviewed BA/BS chemists who have had undergraduate research experience in nanomaterials and atomic force microscopy but could not draw a reaction scheme for the Fisher esterification to form ethyl acetate, much less identify the peaks on 1HNMR. As a former organic assistant prof, I find it sobering and a little unexpected.
A mechanistic understanding of carbon chemistry is one of the keepsakes of a year of sophomore organic chemistry. It is a window into the Ångstrom-scale machinations of nature. The good news is that the forgetful job candidate usually can be coached into remembering the chemistry. After a year of sophomore Orgo, most students are just glad the ordeal is over and they still may not be out of the running for medical school.
I think the apparent lack of interest in industry is because few have even the slightest idea of what is done in a chemical plant and how chemists are woven into operations.
To a large extent, the chemical industry is concerned with making stuff. So perhaps it is only natural that most academic chemists (in my limited sample set) aren’t that keen on anything greater than a superficial view of the manufacturing world. I understand this and acknowledge reality. But it is a shame that institutional inertia is so large in magnitude in this. Chemical industry needs chemists of all sorts who are willing to help rebuild and sustain manufacturing in North America. We need startups with cutting edge technology, but we also need companies who are able to produce the fine chemical items of commerce. Have you tried to find a company willing and able to do bromination in the USA lately? A great deal of small molecule manufacture has moved offshore.
Offshoring of chemical manufacturing was not led by chemists. It was conceived of by spreadsheeting MBAs, C-suite engineers and boards of directors. It has been a cost saving measure that mathematically made sense on spreadsheets and PowerPoint slide decks. The capital costs of expansion of capacity could be borne by others in exchange for supply contracts. There is nothing mathematically wrong with this idea. Afterall, corporate officers have a fiduciary responsibility to their shareholders. Allowing profit opportunities to pass by is not the way to climb the corporate ladder.
We have become dependent on foreign suppliers in key areas who have control over our raw material supply. Part of control is having manufacturing capacity and closer access to basic feedstocks.
The gap between academia and industry is mainly cultural. But it is a big gap that may not be surmountable, and I’m not sure that the parties want to mix. But, I’ll keep trying.
[Note: This post is about replacing the hydrogen atoms along the carbon backbone of a polyolefin polymer with fluorine atoms to produce a fluorocarbon surface on a finished good. Here “finished good” refers to anything from polyolefin pellets, powders, components or blow molded articles such as HDPE bottles.]
Recent news has highlighted the use of fluorinated High-Density Polyethylene (HDPE) packaging for pesticides and other products, bringing more attention to the issue of PFAS/PFOS contamination.
What we’re not talking about is a polymer made from fluorinated monomers or comonomers. This refers to a hydrocarbon HDPE bottle made from ethylene (H2C=CH2) monomer that is fluorinated after the bottle is manufactured.
What’s more, the HDPE fluorination process is said to produce PFAS/PFOS (how?) substances that can migrate. Although this technology is not new, and fluorinated hydrocarbon bottles have been around well before the widespread concern over PFAS/PFOS residues, the significance of such contamination was not fully anticipated. As a chemist, the extensive release of fluorinated low molecular weight alkyl derivatives like PFAS/PFOS came as a surprise to me despite knowing that an analogous situation with fluorinated pharmaceuticals that are getting through wastewater plants due to their resistance to microbiological decomposition. For myself only, very little concern for PFAS/PFOS pollution has been noted. You might suppose that chemists could have led the way to understanding. But, not to my knowledge.
The perfluorinated alkyl materials in question bear a close resemblance to TeflonTM which is known for its chemical inertness and lubricity. In chemistry, Teflon is usually ignored as unreactive with most chemicals, except perhaps molten alkali metals. Strategically placed fluorinated features on a molecule can lend the property of greater hydrophobicity or lipophobicity with increased electron withdrawing properties. The high electronegativity of fluorine pulls electron density towards the fluorine atoms through the sigma bonds of a molecular skeleton. Fluorinated organic acids very often have dramatically increased acidity like triflic acid, CF3SO3H, or increased alkylating reactivity like magic methyl, F-SO2(OCH3). By contrast, fluorinated carbon chains themselves are fairly unreactive and quite hydrophobic, as in water repellant. The water repellency of fluorinated hydrocarbons is a very attractive property commercially.
Below are images of the hydrocarbon hexane in ball and stick form and below in a space filling rendering. To the right is perfluorohexane and below that is its space filling rendering. Hexane is just an example of an “ordinary” hydrocarbon that could be perfluorinated.
Graphics by Sam Hill. Hexane (left) and perfluorohexane (right). As can be seen on the right, the green fluorine atoms are rendered larger than the corresponding white hydrogen atoms because fluorine atoms are larger than hydrogen atoms. In some rendering software, the space filling structures are adjusted to show where some percentage (i.e., 95 %) of the electron density is located. These renderings are by ChemSketch so God only knows how atoms are scaled.
A brief interlude on molecular polarity
Before we go on, there is the matter of polarity, dipolarity, dipolar chemical bonds and dipolar molecules. A dipolar polar chemical bond is one in which the distribution of electrons is lop-sided. That is, one atom of a chemical bond has a bit more negative charge than the other, which is thereby deficient in negative charge, or by default carrying a partial positive charge. Chemical bonds, functional groups and entire molecules can be dipolar.
But charge comes in whole numbers, so how can we talk about partial charge? A covalent chemical bond consisting of 2 atoms, same or different, will hold together because the two atoms share a pair of outer electrons. If one of the two atoms in the bond has a greater affinity for negative charge, then the cloud of 2 bonding electrons will spend a bit more time near the more electronegative atom. This shift leaves the other nucleus slightly deficient of negative charge averaged over time meaning that the positive charge of the nucleus is slightly more exposed to the world.
Graphics by Jed Klampett. Polar and nonpolar molecules.
In chemistry there is a saying- “likes dissolve likes”. This means that a polar solvent like water can more readily dissolve polar solids and may mix freely with other polar liquids. Nonpolar liquids like hydrocarbons can dissolve nonpolar solids and may mix freely with other nonpolar liquids. Amphiphilic substances have both polar and non-polar features allowing them to compatibilize polar and nonpolar molecules together. Soaps and detergents are in this category.
We should be careful here. The polar-polar and nonpolar-nonpolar solubility generalizations above are really just bookends across a vast open shelf of partial solubilities between them. Nonetheless, it is a useful rule of thumb.
So, if likes dissolve likes, and the fluorine atoms on a molecule accumulate a bit of negative charge, then why doesn’t a fully fluorinated organic molecule freely dissolve in water owing to fluorine’s negative polarity via hydrogen bonding with water’s positively polarized hydrogen atoms?
Carbon atoms can form bonds with itself or other atoms in several ways that give rise to different overall shapes.
Back to our regularly scheduled content
In situ fluorinated packaging, a niche within the packaging industry, was not something I was fully cognizant of until recently. I have come to understand that HDPE, along with numerous other polymers, can undergo treatment with elemental fluorine or fluorinated reagents to alter the hydrocarbon polymer’s C-H groups and convert them into C-F groups. This alteration gives the HDPE surface properties similar to a perfluorocarbon like Teflon™. For HDPE pesticide packaging, this fluorocarbon layer reduces the product’s permeability to the pesticide’s components. Package fluorination is all about reducing permeability of the container.
Definition: Hydrocarbon. A category of substances composed only of hydrogen (H) and carbon (C). There are 4 main sub-categories: Alkanes, alkenes, alkynes, and aromatics. A hydrocarbon can be composed of any combination of the 4. The principal mineral sources of hydrocarbons are coal, petroleum and natural gas. A hydrocarbon may also be called an organic substance, where organic refers to a carbon-based substance.
HDPE, high density polyethylene, is a hydrocarbon polymer of ethylene gas and often with various hydrocarbon comonomers. Hydrocarbon polymers, also called polyolefins, are notable for their considerably inert chemical properties. Inertness is the resistance to chemical change. However, contact with certain fluorinating agents like F2, ClF3, NF3, etc., diluted in an inert gas can, at relatively low temperatures, exchange the H atoms of HDPE with F atoms. Eventually, all or most of the H atoms on the polymer surface will be exchanged. A carbon molecule that has F atoms replacing all H atoms is said to be perfluorinated.
Pesticides are meant to be spread over selected parts of the environment to do their trick. A great many pesticides are synthetic organic chemicals so naturally there is the possibility of any given pesticide or solvent to diffuse through a hydrocarbon-based container. Migration of product molecules into the polyolefin packaging, in this case (HDPE), can result in the release of the hazardous contents and compromise the overall containment, possibly resulting in exposure to the public and the environment.
It should be possible to slow the rate of diffusion of any given hazardous material through a non-fluorinated container by simply making the container walls thicker. The polyolefin manufacturers would be in favor of this, but the converters who buy the plastic pellets to blow mold the containers may balk. Their raw material costs would rise and they would have to pass the costs to customers, who will resist the cost increase. Then with the increase in mass flow of polymer melt necessary, perhaps the throughput or required extruder torque might change unfavorably. Hard to say.
Some of the small-molecule bad actors
On March 5, 2021, EPA published the list below of PFAS/PFOS compounds found in the 20-50 ppb level in fluorinated HDPE containers used to store and transport a mosquito control pesticide product.
Abbreviated
Full Name
PFBA
Perfluoro-butanoic acid
PFPeA
Perfluoro-pentanoic acid
PFHxA
Perfluoro-hexanoic acid
PFHpA
Perfluoro-heptanoic acid
PFOA
Perfluoro-octanoic acid
PFNA
Perfluoro-nananoic acid
PFDA
Perfluoro-decanoic acid
PFUdA
Perfluoro-undecanoic acid
These are all perfluoroalkyl carboxylic acids listed by increasing chain length. Notably the terminal carbon is fully oxidized to the carboxylic acid and is not fluorinated. This acidic end gives a chemically reactive handle for further manipulation of the PFAS/PFOS if desired.
PFOA, perfluorooctanoic acid, has been industrially produced by what is now 3M since the mid-1940s. It has been used to place TeflonTM coatings on frying pans. It was originally prepared by the electrochemical fluorination (ECF) of octanoyl (ock TAN oh ill) chloride, the hydrogen saturated 8-carbon acid chloride. ECF produces the perfluorooctanoyl fluoride which is then hydrolyzed to the acid chloride liberating HF.
Perfluorination of HDPE bottles relies on the most electronegative element, diatomic fluorine gas, F2, or other similarly reactive fluorinating reagents, and does chemistry on a solid polyolefin surface. Fluorine gas is diluted in a suitably noninterfering gas like nitrogen, argon or CO2 and then exposed to the polymer of interest at a prescribed pressure, temperature and exposure time. Fluorine atoms replace hydrogen atoms on the polymer chain. According to one source, the rate of fluorination is diffusion limited. This means that the fluorination reaction is very fast. The presence of molecular oxygen with molecular fluorine had a retarding effect on fluorination proportional to the concentration of oxygen gas. The presence of oxygen led to it being incorporated onto the polymer.
Given the advantage of impermeability provided by fluorinated polyolefin articles, it is clear that there are many excellent applications of in situ fluorinated polyolefins. The replacement of glass and metal with lighter fluorinated HDPE containers may save on transportation costs on a weight basis. Whether or not the economics favor fluorinated polyolefins over glass or metal manufacturing costs kg for kg is unclear.
The range of application categories listed above is quite large. Each entry in the list has many individual components that may be subject to fluorination as well. It is no wonder that PFAS contaminants are spread widely around the world. The US EPA has issued a letter (below) to companies fluorinating HDPE to beware of accidentally producing PFAS/PFOS in their operations. Specifically warning about the connection of PFAS formation caused by the inclusion of oxygen in the fluorination process. The letter specifically cites “EPA’s 2020 long-chain perfluoroalkyl carboxylate (LCPFAC) Significant New Use Rule (SNUR) (40CFR § 721.10536), that are found to be present in or on fluorinated polyolefins may be subject to TSCA regulations and enforcement.”
“It is during certain types of fluorination (e.g., the presence of oxygen) that the manufacture of PFAS has occurred. Manufacturers (including importers), processors, distributors, users, and those that dispose of fluorinated HDPE containers should be reminded of this potential for manufacturing PFAS and comply with any applicable regulations under TSCA, as described in the next section.“
“EPA is aware of alternative fluorination processes that use fluorine gas in the presence of gaseous inerting (e.g., nitrogen) without the presence of oxygen that could reduce the potential for unintentional manufacture of PFAS. These alternative processes for fluorination of polyethylene are highlighted in the U.S. Food and Drug Administration’s (FDA) August 2021 letter on this issue as it relates to food contact articles.”
“Requirements under TSCA PFAS Significant New Use Rules. Certain PFAS, including long-chain PFAS as defined in EPA’s 2020 long-chain perfluoroalkyl carboxylate (LCPFAC) Significant New Use Rule (SNUR) (40 CFR § 721.10536), that are found to be present in or on fluorinated polyolefins may be subject to TSCA regulations and enforcement. EPA considers the manufacturing of certain PFAS from the fluorination of polyolefins to be a significant new use under TSCA. LCPFAC chemical substances present in polyolefins due to the fluorination process would be considered byproducts of the manufacturing process because they are produced during the manufacture of the fluorinated polyolefins and do not have a separate commercial intent (40 CFR § 720.3(d)). LCPFAC chemical substances that are byproducts of the manufacturing process for fluorinated polyolefins do not meet the requirements of the byproducts exemption at 40 CFR § 721.45(e)5 and are subject to significant new use notice requirements. Significant new use rules require industry to notify EPA at least 90 days before commencing the manufacturer including import or processing of subject chemical substances for a significant new use. The required significant new use notification (SNUN) initiates EPA’s evaluation of the conditions of use associated with the significant new use. Entities may not commence manufacturing (including import) or processing for the significant new use until EPA has conducted a review of the notice, made an appropriate determination on the notice, and taken such actions as are required in association with that determination. Tala R. Henry, Ph.D., Deputy Director Office of Pollution Prevention & Toxics 2022/03/24.”
Fluorination and fluoridation. What’s the difference?
So we do not make people worried about their fluoride toothpaste or their fluoridated drinking water, let’s sort this out. Toothpaste and drinking water have a soluble ionic fluoride salt like sodium fluoride, NaF, or sodium monofluorophosphate, sodium MFP or chemically Na2PO3F. Sodium MFP is water soluble but not stable in water. It hydrolyzes to release fluoride by displacement by water to form dibasic phosphate. The MFP hydrolysis reaction is: PO3F2− + HO– → HPO42− + F−. The fluoride anion, F–, is not nearly the same as fluorine gas, F2. The F– ion bumps into tooth enamel where it binds tightly with calcium in the tooth: Ca5(PO4)3+(aq) + F−(aq) → Ca5(PO4)3F(s). This is the context in which the word “fluoridation” is used. Fluoride ions bond tightly to calcium++ ions in general. Fluoridation is just a specialized variety of fluorination and is mostly confined to the area of water treatment and toothpaste.
Fluorination is a chemical process wherein fluorine atoms are added to chemical compounds. Contact between organic substances and pure elemental fluorine gas is extremely exothermic and sometimes explosive. The dilution of F2 gas with an inert gas like nitrogen, helium or argon has a thermal safety component as well.
Polymer fluorinationout in the world- Patents
One source of manufacturing information about proprietary articles and processes is the US Patent and Trademark Office, USPTO. In order to secure your legal right to a patent, the patent applicant must disclose the exact art that is being claimed. This is because the world must have a fair chance to avoid infringement. Google Patents provides the exact text of individual patents, US and others. It also provides a timeline showing the ownership of the patent and whether or not the patent is active, expired or abandoned. Google patents also provide links to patents cited in the patent and patents that have cited the instant example.
Being a Google product, Google Patents has extensive and flexible search capacity. Rather than attempt to make a list, it is a better use of the reader’s time to go to the site yourselves and explore. Note that a search will find patents from all over the world as well as patent applications. Google patent provides a English translated version of the patent.
In searching for patents claiming compositions and methods around the fluorination of polymers, more than a few patents can be found. One can search for patents using the USPTO website (obviously) or from Google Patents.
Another good place to look for relevant art is from a patent you have already pulled up in Google Patents. Near the bottom of the patent from Google Patents is a section labeled “Patent Citations.” This section list prior art patents disclosed by the assignee and those found by the patent examiner in the course of the examination process. Prior art is disclosed by the assignee in the granted patent as well, but in Google Patents there are hotlinks to patents to aid the convenience factor.
In situ fluorination
There are companies who will fluorinate the surface(s) of High-Density PolyEthylene (HDPE) and PolyPropylene (PP) containers. HDPE and PP are especially of interest owing to their utility in packaging liquids. These two polymer classes have great rigidity and strength and are in wide use. However, they share certain weaknesses such as air permeability and permeability of the contents. Air permeability is highly undesired in food packaging as it allows for reduced shelf life or customer satisfaction with the contents. Food and drugs may be susceptible to air degradation and possible reduction of shelf life.
Note: The expiration date on a product does not necessarily mean that the product will go bad when that date arrives. The day after the expiration date is that date at which the manufacturer/seller will no longer guarantee “freshness” or some other type of quality. For instance, normal pasteurized milk is not sterile. Pasteurized milk should be good up to 1 week past the code date as long as it has not been allowed to warm up or been contaminated. Once the milk has warmed to room temperature, the normal bacteria loading will enter log-phase growth and could spoil within 1 day.
In situ fluorination is process wherein hydrocarbon polymer containers are exposed to diluted fluorine gas at a specified temperature for a specified time. At the surface hydrogen atoms along the length of the polymer are replaced with fluorine atoms. The result is a polymer along the surface which resembles TeflonTM to some extent. Some of the desirable properties of TeflonTM are then taken on by the HDPE or PP surface. This H/F exchange at the surface does not affect the properties of the base polymer.
There is one caveat, however. The fluorination must be performed with the exclusion of oxygen. One source says that the vacuum chamber in which the fluorination will take place must be pumped down to 0.1 Torr of residual air prior to exposure to fluorene gas.
Fluorination patents
Below us from the description in US5274049A Filing date 1991-07-19, Application filed by SHAMBAN WILLIAM S, W S SHAMBAN AND Co.
A method for the direct fluorination of elastomers “in order to reduce the static and dynamic friction characteristics and to increase the wear life and abrasion resistance of the elastomers. The invention also relates to elastomeric articles modified by the fluorination method.”
“What is claimed is:
1. A method of producing fluorinated elastomeric articles, consisting essentially of the following steps:
providing an elastomeric article, said elastomeric article comprising an elastomeric polymer having a backbone chain having a plurality of hydrogen atoms attached thereto;and
exposing said elastomeric article to gaseous fluorine under conditions sufficient to reduce the friction coefficient of said article without promoting degradation of the tensile properties of said article.”
Claim 8 claims a method using a hydrogen fluoride scavenger …
“8. A method for producing a fluorinated elastomeric article having a reduced coefficient of friction, comprising the steps of:
placing a thermoset elastomeric article and a hydrogen fluoride scavenger in a closed reactor vessel, said thermoset elastomeric article comprising an elastomeric base polymer having a backbone chain, said backbone chain including sufficient carbon atoms having replaceable aliphatic carbon-hydrogen bonds so that a fluorinated matrix of said fluorinated elastomeric article reduces said coefficient of friction;”
In the description the patent cites sodium fluoride, NaF, as an HF scavenger wherein NaF + HF => Na[HF2], sodium bifluoride.
Inhance Technologies LLC filed application US20190040219A1, but it was later it was abandoned due to failure to respond to an office action. The application claimed a multistep method for fluorinating elastomeric workpieces with 20 % F2 in nitrogen and “altering certain mechanical properties such as tensile property [and] the elastic modulus, an impact property, a wear property, etc.“
Systems and methods for processing fluoropolymer materials and related workpieces, US11879025, filed 2021-04-23, Current Assignee: Inhance Technologies LLC. Claims method of removing perfluorinated compounds from fluoropolymers. The core of the art involves placing a fluoropolymer work piece in a thoroughly deoxygenated chamber, heated from 25 C to 300 C and exposed to a fluorinating atmosphere such as F2/N2 for specified time period. This treatment is claimed to remove fluorocarbons like PFOA to non-detectable levels. There is no mention of where the PFOA goes afterwards, but it looks promising if accurate. However, the granted patent is off-limits for 20 years unless a license is obtained or some other arrangement is made.
Fluorination is imbedded deeply into the design of a great many articles of commerce. The water repellency of perfluorinated polymers in fabrics is one of the chief applications of fluorinated organic materials. The inherent lubricity of PTFE, its built-in chemical inertness and its hydrophobicity have ingratiated millions of consumers and have met performance expectations world wide.
Perfluorinated foams for fire protection in aircraft hangers and industrial spaces are valuable for their ability to float on the surface of burning liquid fuels, blanketing the surface as a vapor and oxygen barrier. The suppression of flammable volatiles in a fire by a layer of protective foam can inhibit flashover of the fire, reducing the overall damage of a fire. The fire retardancy of perfluorinated substances inhibits their combustion and discourages continued burning when the flame source is removed. Halogens as a group have been used for fire retardancy and with bromine in particular.
The chemical origin of the fire retardancy properties of perfluorinated organic materials lies in the low reactivity of the -CF2– fluorine atoms with oxygen. In the combustion of hydrocarbons, hydrogen atoms are readily removed by oxygen or radical species to form water. The C-F bond is one of the strongest bonds in organic chemistry and is slow to be removed by oxygen.
Drug molecules are frequently fluorinated in particular locations on the drug molecule. A C-F bond resists catabolic degradation and enhances the local hydrophobicity of the drug allowing for greater half-life and enhanced drug potency. The down side is the resistance to catabolic degradation and excretion. Many drug molecules are released intact into sewage treatment facilities where they also resist degradation, possibly due in part to the fluorinated features. The effect is that fish and other organisms are exposed to the drug. As with humans, fish and other creatures of the waterways and soil did not evolve with biochemical mechanisms to deal with fluorinated organics.
In the in situ fluorination process, PFAS/PFOS side products can form, especially when oxygen is present. This can be monitored by quality control but companies will comply with recommended PFAS/PFOS best practices only if there are regulations or the threat of them. Nations regulating PFAS/PFOS contamination will have to compete with nations who do not impose regulations. This is the usual scenario for nations with heavy reliance on imported articles but uneven regulation.
The state of Nevada is quickly becoming the leading source of lithium in the USA and beyond. In the state there will soon be three major types of lithium ore beneficiation- Brine evaporation, hard rock extraction and lithium clay extraction. Nevada already has in excess of 180,000 active mining claims amounting to 49 % of the total BLM national inventory. In addition to this, Nevada has “198 authorized mining plans of operations, and 282 active exploration notices.” Nevada has a long history of fruitful gold and silver mining.
Nevada had earlier won the gold deposit lottery with the Carlin Trend occupying much of the northwestern section of the state. The Carlin Trend has become an archetype in gold mining. These deposits are often described as Carlin-type “invisible gold” ore deposits. Such a deposit is characterized as sediment hosted and disseminated [Editor: disseminated seems like a bummer]. Gold in such deposits are typically invisible and often only detected by lab analysis. According to Wikipedia, most of the gold mines in the Great Basin of the western US are of the Carlin-type.
But, enough about gold and on to lithium
After 6 years of regulatory scrutiny, a new lithium-boron open-pit mining operation in Nevada operated by Australian mining company ioneer has just been approved by the Bureau of Land Management, BLM, for Rhyolite Ridge. The mine is located in the Basin and Range Province near the southwest border of Nevada and California.
The old joke used to be that a mine is a hole in the ground with a liar standing at the top. With the independent economic evaluations available today as part of the disclosure to investors, the likelihood of being duped by fake or salted deposits has dropped considerably. However, the market value of the ore in the future is still subject rapid and unpredictable change.
If you find yourself flying over Nevada on a clear day, you can easily see the basin and range features of the terrain. Nevada occupies only a small part of the total area. The basin and range province extends north to the Columbia Plateau and south into the Central Mexican Plateau.
A very small part of Nevada’s basin and range landscape as viewed from above the Rhyolite Ridge area in Nevada. Image from Google Maps.
The Basin and Range Province of North America. Image from Wikipedia.
Rhyolite Ridge Lithium-Boron Project
The BLM approval opened up $1.19 billion of potential funding of which $700 million is from a US government loan. According to Mining.com, Rhyolite Ridge is the first new lithium mine in 60 years and the first new boron mine in the last century in the US. [Note: I have to assume “new” means new hard rock mineas opposed to brines or evaporites] While the approval by BLM has opened some doors to funds, not everyone is convinced of the major investor’s liquidity.
So, what is rhyolite?
I can’t improve on the definition found in Wikipedia, so I’ll just quote it with the links intact-
If you have ever seen molten glass and noticed its high viscosity, this gives an idea of what high silica content does to lava. The higher viscosity provided by the silica component suppresses the release of gases until nearer the surface where they are released as bubbles with vigor. It is very much like a comparison between boiling pasta water and boiling marinara sauce. The marinara sauce spatters badly due to its viscosity but the pasta water just does a rolling boil.
Source: Mashed.com. Spattering is a universal behavior of hot, gassy fluids. In this case the gas is steam. Magma also contains steam.
The Rhyolite Ridge lithium-boron (LiB) deposit is said by some to be the only known LiB deposit in the US and only one of two known in the world.
“The Rhyolite Ridge mine will run for 22 years. It will produce 22,000 tonnes of lithium carbonate a year. That’s enough to power 370,000 electric vehicles. It will also produce 170,000 tonnes of boric acid, according to the company. The boron contributes 30% to 40% of the mine’s revenue, providing a buffer against lithium market volatility.” –Mining.com
Around the world new economic lithium deposits are being discovered now and then, and a few are being readied for mining. It was announced recently that BLM has approved operations at the Rhyolite Ridge Lithium-Boron Project in southwestern Nevada.
What is interesting about this Rhyolite Ridge project is that it aims to produce both lithium and boron. I’m not an engineer so maybe I’m overly impressed, but the processing plant they propose seems very clever. They will produce their own sulfuric acid from sulfur and extract waste heat for use in generating steam for evaporation of the extracts and electricity. They are completely off the energy grid.
The extracted ore, now called ROM or run-of-mine, is transported to the plant straight from the mine and sized by crushing to 20 mm pieces. The crushed ROM is then taken to a series of sulfuric acid extraction vats and leached for ~ 7 days. The pregnant leach solution containing the lithium, boron and soluble impurities is then taken to evaporators with repeated crystallizations and, using differential solubility, separates the lithium component from the boric acid. In the end they produce lithium carbonate. The video does show a soda ash (sodium carbonate), Na2CO3, silo so I assume that is where the carbonate comes from to produce lithium carbonate, Li2CO3 and to neutralize residual sulfuric acid.
Silver Peak Lithium Brine
Of interest is the nearby Silver Peak lithium brine operation operated by Albemarle just a few miles to the north of Rhyolite Ridge. The Google Maps image below shows the evaporation ponds at the Silver Peak lithium operation. Silver Peak produces both technical grade lithium carbonate and lithium hydroxide.
Image from the Operational Land Imager-2 (OLI-2) on NASAs Landsat 9. A view from space of the Silver Peak lithium brine evaporation ponds in SW Nevada.
McDermitt Caldera: Thacker pass
Another large lithium deposit was discovered in the McDermitt Caldera along the Nevada-Oregon border. Within the caldera is the Thacker Pass Lithium Mine. This lithium deposit was approved for open-pit mining by BLM on January 15, 2021, though it has been plagued by protests and an injunction. As with the rest of the McDermitt Caldera lithium, the Thacker Pass lithium is described as a lithium rich clay deposit. This is unique for lithium mines since brine extraction and hard rock mining of spodumene have been the norm. Thacker Pass’ lithium deposit is the largest known volcano sedimentary deposit in the US at an average grade of 0.22 %.
In 2023 GM invested $650 million in the Canadian Lithium Americas Corp. The Thacker pass operation is through its wholly owned subsidiary Nevada Lithium, LLC, which is responsible for production. Car giant GM’s investment gives them exclusive access through the first phase of production. Lithium Americans has received a conditional approval for a $2.2 billion loan from the US Department of Energy.
The Thacker Pass measured and indicated lithium resources are 13.7 million tons of lithium carbonate equivalent. Lithium Americas calculates that the recoverable lithium is worth $3.9 billion.
Interestingly, the McDermitt Caldera is possibly the oldest of a sequence of calderas produced by the Yellowstone Hotspot. The McDermitt Caldera amounts to a lava dome that collapsed ~16.4 million years ago forming a large caldera within which several smaller calderas have formed and in which later filled with water forming a lake over the tuffaceous ash. Over time the lake produced sediments that were deposited on the floor of the lake. The source rock is rhyolite which is usually the case in the state.
The Yellowstone hotspot stays relatively constant while the crust moves over it, leaving a trail of calderas and a record of volcanic activity on the surface behind. The McDermitt caldera is labeled ’16’. Source: National Park Service.
Briefly, the lithium clay is excavated, gravel and rocks are removed, and the clay is suspended in water to form a slurry. The slurry is leached by adding sulfuric acid that is produced on-site and the lithium in the ore is extracted into the acidic liquor. Finally, the dissolved lithium is recovered as lithium carbonate and lithium hydroxide. Gangue material is deposited back into the excavated sections of the mine.
The McDermitt caldera contains many breccia and fracture zones and associated with these are deposits of other metal ores. Specifically, mercury from cinnabar ore bodies and uranium from autunite (uranyl, U6+) (Ca(UO2)2(PO4)2·10–12H2O) and pitchblende (uranate, UO2, U4+) ore bodies. Mercury reserves in the caldera, according to USGS, are estimated to be 400,000 flasks.
A Mercurial Rambling
Mercury has been packaged (and still is) at 76 pounds to the flask. This measure got its name from the mining and smelting of cinnabar in the mountains of Peru. One site was particularly rich- Huancavelica, Peru. The Spaniards had known prior to their arrival to the New World that liquid mercury could dissolve gold from ore to produce what we now know as a mercury amalgam. With very strong heating the mercury could be driven off as vapor to recover the precious metal.
Spain had its own cinnabar mine in what is now Almadén, Spain, which produced over 250,000 tonnes of mercury. The Spaniards had considerable experience with mining, refining and using mercury prior to discovery of it in the new world. Mercury was quite valuable to the Spaniards but they were faced with transporting it from, say, Huancavelica, Peru, to the Carribean coast where their ships could load the mercury and distribute elsewhere. Anecdotally, it has been estimated that for every ounce of gold produced in the new world, ten ounces of mercury were consumed.
Luckily for the 49er gold rush miners, cinnabar had previously been discovered in California. You can actually visit the old New Almaden mine museum south of the Bay Area. It is worth a trip if you are in the area.
The Spaniards figured that a man could carry what turned out to be as much as 76 lbs of mercury through difficult terrain to the Caribbean coast. Even better for the Spaniards, they knew how much gold could be extracted per pound (or whatever unit) of the mercury they dispensed. This gave them an idea of how much gold to expect and closer control of the mines. By controlling mercury, they controlled who could use it for mining and how much gold they could recover.
Back to lithium
As of this writing, Albemarle is the largest producer of lithium in the US. But the largest known deposit in the US is at Thacker Pass. The Albemarle Silver Peak lithium brine operation is legally defined as placer mining whereas the Thacker Pass operation is lode mining. The word ‘legally’ is used because any claims filed are restricted to placer or lode mining- one does not transfer to the other.
A Word or Two About Rhyolite
Rhyolite is a type of volcanic rock characterized as a ‘fine-grained extrusive igneous rock‘. Its color can vary from pale light grey to pinkish when composed of mainly quartz and feldspar or dark when of a mafic, low silicate composition was present. A quiet eruption of lava gives a solid, denser rhyolite whereas in an explosive eruption can produce the vesicular pumice. The lower density of pumice allows it to float on water. Erupting volcanic islands can produce floating rafts of pumice in the nearby waters. As magma rises in the throat of a volcano, the pressure drops and dissolved gases can form bubbles which, if they fail to disengage from the magma completely, can be ejected into the cooler air and freeze into ‘foamy’ structure. [Note: the word ‘foamy’ is my own and earth science people cannot be blamedfor this.]
A Bit More About McDermitt Caldera
The United States Geological Survey (USGS) published open-file report 76-535 in 1976 titled Geology and Ore Deposits of the McDermitt Caldera, Nevada-Oregon. A 1978 USGS open-file report by Newmont Exploration, Ltd., 78-926, James J. Rytuba and Richard K. Glanzman, Relation of Mercury, Uranium and Lithium Deposits to the McDermitt Caldera Complex, Nevada-Oregon, goes into greater detail on the three minerals.
What can a chemist possibly have to say that could be even marginally interesting about extraterrestrial life or evolution? Well, as far as extraterrestrial life and the search for it goes, I would say that all of the metallurgy, semiconductor fabrication, liquid hydrocarbon fuels, chemicals, transportation technology or polymers exploited in radio or optical astronomy, have some element of chemistry in their manufacture.
………………..
The quest to discover life beyond Earth captivates many in the broad field of space science. The Search for Extraterrestrial Intelligence (SETI) has played a significant role in astronomy and space science communities. However, the search extends beyond intelligent life; any form of life or even the essential components and conditions conducive to life, are of keen interest.
It is widely accepted that the physics governing our planet and solar system likely applies universally. While this is a hypothesis, it is a reasonable one. If the physics are consistent, then the chemistry should be as well. Consequently, the behavior and limitations associated with matter would be uniform across the cosmos. This reasoning suggests that life elsewhere in the universe would be governed by the same chemical and quantum mechanical principles familiar to us.
The “Anthropic Principle” has caused much debate, with Wikipedia noting that “Anthropic reasoning is often employed to address the notion that the universe appears to be precisely calibrated for life.” The mystery of why numerous physical constants and their ratios needed to be exactly as they are for life to emerge on Earth has intrigued many.
To say the Big Bang’s initial pressure and temperature were high is an understatement. As the universe expanded and cooled, energy barriers emerged that shaped the interactions of matter and of photons, placing boundaries on the spontaneous transformation behavior of matter. Pathways of interaction emerged, steering transformations towards increasingly specific outcomes. Essentially, it’s basic kinetics: the quickest transformations and their stable products start to prevail and fill the universe.
If physical constants are emergent at the moment of the Big Bang and become manifest down the timeline, could it be that another Big Bang could happen that is not conducive to life? There would be nobody there to ponder these questions. Life is here because it was possible and maybe even likely here and there.
The phrase “finely tuned for the existence of life” seems to leave open a crack for a creationist view. Absent the many spooky bronze and iron-age theories still in practice today, naturally a sentient being can look at her/his/its existence and marvel at how beautifully synchronized and proportioned the machinery of the universe is. Certainly, there must be a hidden message in this, right?
ET? What th’ …?
Animals like mammals, birds, fish, and even some invertebrates like octopi and crabs are considered to be sentient. According to Google, sentient animals are those that can experience feelings and sensations, both positive and negative, like pleasure, pain, joy, and fear. So, while an octopus may have elements of sentience, could distant observers elsewhere in the galaxy detect them from optical or radio astronomy techniques? Try as it might, the ability of an octopus to construct a powerful radio transmitter and beam a message into the cosmos is sorely limited by its physical anatomy. Except for humans, no other sentient life form on Earth is known to construct a radio transmitter that would serve as a beacon of sentient life.
Until recent history, SETI was limited by the lack of technology to light up the universe with our own signals or to detect faint manufactured signals across interstellar space. At such point that metallurgy, electrical engineering and the hundreds of other critical and apex technologies bloomed into a sufficient state of development, no intelligent emanations from Earth found their way into space.
While TV and radio broadcasts began their journey into space, it is important to realize that our signals were encoded onto carrier waves. Amplitude modulation (AM) signals carry their information by simply varying the magnitude of a single frequency in time with the human voice or music. This is most likely to be grasped by alien radio astronomers. Frequency modulation (FM) is a bit more challenging because audio signal is mixed with a carrier signal by a heterodyne circuit. Extracting useful information would require them to pull audio frequency information from the heterodyned signal.
Television is much more difficult. While the alien radio astronomers may have figured out FM encoded radio information, the particular details of the TV raster scan are based how human engineers decided to interlace and sequence scans to produce an image on a screen of a particular aspect ratio. TV designers took advantage of the human’s persistence of vision to seamlessly follow moving pictures to give continuous images yet maintaining a fast enough frame rate to avoid flickering. The television’s electronic timing is based on frame rate, the number of interlaced lines, and the aspect ratio of the screen.
The point of this TV discussion is that a TV signal must be deconvoluted into a signal that properly displays an image and plays the sound on a particular piece of equipment. This could be challenging for an alien radio astronomy research group to decode.
All of this talk about an octopus developing radio astronomy presupposes that its unique octopussian sentience includes such desires.
It could be that the initial energy at t = 0 yielding the primordial plasma constituting the early Big Bang was only capable of producing a specific set of fields producing elementary particles which then give way to a specific set of quantitative relationships and properties. The burst of energy causing the Big Bang must have had constraints driving its transformation into matter, which is also constrained by quantum mechanics, etc. Maybe the present universe is simply what primordial energy naturally does when expanding as a universe. Why do the quantitative values of physical constants need to be variable? An imaginary and feverish conundrum.
As the highly energized primordial plasma of the Big Bang began to cool, matter and energy channeled into particular states. The particle energy states that had the highest barriers coalesced first followed by subsequent lower energy plasma condensing into other particles. I’m drawing a crude analogy to the process where individual minerals form from cooling magma according to their melting points.
There is a notion prevalent among Creationists that the probability of a life form spontaneously forming from individual atoms is 1 in 10large or some other inconceivably miniscule chance. And if that was how life had to form, then the Earth would still be a sterile wet rock. But that is not how chemical transformations work.
Central to the Creationist view is that evolution cannot happen because there is nothing but random chance to guide the molecules of life into a highly complex organism. They start with the assumption that life arose purely from random chance. I hope to show that this assumption is false.
All atoms and molecules have properties that either qualify or disqualify them as a candidate for a given atomic or molecular transformation. All molecules have properties that either qualify or disqualify them to take part in a transformation resulting in a given product. The words “qualify” or “disqualify” could mean that something will or will not happen absolutely. But just as likely, the words could mean that a transformation is just too slow at a given temperature to give the desired effect. As it happens, temperature is critically important to molecular transformations. At a low enough temperature, most transformations will slow to a negligeable rate, shutting down that particular transformation channel. In general, where there are competing transformation channels, the fastest channel will prevail in producing its product.
All molecules have a limited set of reaction channels at a given temperature as a result of their particular reactivity.
What we think of as ‘ordinary’ chemistry is more precisely the electronic behavior of valence electrons. Nuclear chemistry also exists but in the domain of nuclear change.
Valence electrons on earth will behave the same everywhere in comparable conditions. Chemistry happens at the outer, valence level of ions, atoms and molecules. So, we should expect that bond forming and bond breaking mechanisms should be the same throughout. All of this leads to the high likelihood that chemical reaction mechanisms elsewhere in the universe should not be unfamiliar to Earthlings in general.
Life on earth exists as a result of the behavior of particular chemical substances within a range of chemical and thermal environments. The range of chemical environments and substances present during the initiation of life is thought to be quite different than what we find on earth at the present time. For instance, gas phase molecular oxygen was not present until a considerable time after life began. The initiation of life on earth was under anaerobic conditions and was able to start and survive with the materials at hand. Biochemistry is a series of reduction/oxidation events driven by the Gibbs energy of a transformation as is all of chemistry. Even on anoxic earth, diverse oxidizers were present.
Today, anaerobes are known to use the oxidative properties of inorganic species like sulfate (SO42-), nitrate (NO3-), ferric iron (Fe3+), carbon dioxide (CO2) and manganese (Mn 4+). Other anaerobic oxidants include chromate (CrO42-) and arsenate (AsO43-) which may have been present as well. Reductants include nitrite (NO2-), ferrous iron (Fe2+), and sulfide (S2-).
Oxygen is the third most abundant element in the universe and the second most abundant heavy element on earth behind iron. Many elements are strongly attracted to the abundant oxygen so it is no wonder that so many minerals are oxides of one sort or another. Oxyanions like silicates, carbonates, sulfate, nitrate, and oxides like CO2 or any number of metal oxides all contain oxygen that has been bound with another element. The oxygen pulls negative charge away from the central element making it electron deficient. In the case of sulfate and others, the actual oxidizing part is the atom with the oxygens attached, in this case the sulfur.
The presence of life on Earth means that there is a “habitable zone in parameter space“. All of the parameters affecting biochemistry must align in such a way that a zone of allowable chemical and physical conditions will exist. Many things must exist simultaneously such as the many properties and abundances of chemical substances, a suitable atmospheric composition and pressure, a planetary temperature range allowing for the presence of liquid water and the presence of sufficiently reactive organic molecules.
Not every transformation of matter is within reach in a given condition. Chemical reactivity which comprises kinetics and thermodynamics has the effect of channeling matter into a finite number of probable pathways. This bestows the property of selectivity. For any given chemical substance, only a certain limited group of transformations are possible or likely, given the conditions.
Life as we know it exists because our biomolecules were robust enough to survive their chemical and thermal environments, but not so robust that they resist the needed transformations. Life depends on biomolecules being moderately stable but not by too much. Biomolecules can organize into particular structures that are physically robust, like the chitin shells on shellfish. In the chemistry of life, chemical transformations must be tolerant of the aqueous environment in and around an organism, but not so tolerant that the necessary reactions are too slow or too fast within the narrow range of environmental temperatures available.
Organisms on earth are tolerant of water at the level of molecules. The internal apparatus of the cell is an aqueous environment having some amount of viscosity. In order for molecules to interact, they must collide with each other. Life in the solid phase would mean that biomolecules would be immobilized and unable to collide and react. Cell structure for metabolism and reproduction would not be feasible. Life in the gas phase is limited by the vapor pressure of the necessary substances. Many, if not most, biomolecules would not tolerate the heat necessary to volatilize. They would decompose.
A diversion into molecular evolution.
I’ll just blurt it out- ongoing evolution requires heritable change in a genome. A genetic change must be survivable for the parent cell to reproduce and produce viable daughter cells. The inherited mutation must not be deleterious to further reproductions of the subsequent generations. A mutation may randomly result in something that has either a lethal effect, no effect, or produces some biomolecular improvement. The mutation may be as modest as an enzyme alteration causing it to bind either more or less tightly to a ligand resulting in a few percent change in rate of some the enzyme’s function. This could translate into better efficiency in producing some cell structure or better use of energy. It could also be that nothing changes as a result of the efficiency alteration, or that it has an overall negative effect further challenging the survival of the cell line in a nonlethal way.
There are two kinds of changes that can occur with DNA. One is a change in the sequence of the DNA molecule itself. The other kind is “epigenetic” which is heritance not reliant on changes on the DNA sequence.
Creationists like to make a show of the probability of random chance producing even simple ordered sequences as fantastically small. Actually, their superficial analysis of permutations and probability looks plausible. I can’t argue with the low probability of individual atoms coming together randomly to form a living organism all at once. However, the beginning assumptions are wrong. Life did not spontaneously form out of a bunch of loose atoms by simply condensing into a centipede or a human. Change in evolution happens at the molecular level a step at a time. A change in the amino acid sequence of any given enzyme must trace back to a change in the DNA sequence to pass along a heritable mutation. Evolution moves by fits and starts. A mutation may have no effect, advantageous effect or deadly effect.
At the level of molecules, change happens through very definite chemical mechanisms. Molecules are constrained to do certain things and in a particular way. It’s like a channel. Sometimes two or more channels may be possible. In this case, the fastest channel will dominate in output and influence. An evolutionary change might cause a biochemical transformation to stop, speed up, slow down, or be more or less specific in outcome.
Molecular bonds vibrate in the range of 1013 to 1014 Hertz. A hydrogen molecule will reportedly undergo 2.5 x 1010 collisions per second at 2 bar and 24 oC. If two atoms or molecules are to react, then they must collide. At a given temperature, a collection of hydrogen atoms will be dispersed over a statistical distribution of energies.
Biochemistry on earth has evolved around water and takes advantage of certain properties of water. Its ability to hydrogen-bond is exploited extensively in biomolecule structures. Water has the ability to accommodate charged species or neutral dipolar species. This is called hydrophilicity. It is important not just to keep ions and molecules in solution, but also to stabilize the transition of a reaction if it generates a momentary dipole.
Water is immiscible with substances having a large hydrocarbon protuberances like fatty acids, phospholipids or certain side groups found on a few amino acids. This is called hydrophobicity. Terrestrial biochemistry exploits both hydrophilicity and hydrophobicity.
A benefit of hydrophobicity in biochemistry is that fatty substances like phospholipids will spontaneously organize into the lipid bilayer structure. Hydrophobicity in this case leads to the formation of stable compartmental structures. Life takes full advantage of the lipid bilayer in the production of the cell wall. This keeps all of the necessary biomolecules contained and concentrated for effective and timely biochemical transformations to occur. The cell wall excludes a great many deleterious substances as well. However, many protein structures have sections that are sufficiently hydrophobic as to be compatible with the hydrophobic lipid part of the bilayer. This property allows the protein to anchor itself within the bilayer leaving the more hydrophilic portions of the protein jutting out into the extra- and intracellular aqueous media. Many of these proteins penetrating the bilayer- channel proteins- are sufficiently hollow as to allow ions or molecules to pass through. Even better, the ability to pass ions or molecules through can be switched on or off by other biomolecules or with drugs.
Some larger molecules like the fatty phospholipids above have both hydrophilic and hydrophobic regions. Given the chance, phospholipid molecules will spontaneously orient themselves in a way that when combined the water ‘repellant’ hydrophobic tails will tend do aggregate. This leaves the hydrophilic phosphate features at each end to remain in contact with the water environment.
Cells have compartmentalization and cell walls simply because of the incompatibility of the polar water molecule and nonpolar hydrocarbons. These two incompatible liquids arrange in a way that minimizes the surface area of contact between them. They will form layers when stationary or droplets when one is dispersed in the other. This is the minimum energy condition they spontaneously go to. Micelles will even form spontaneously in your soapy dishwater.
Life on earth presently requires many environmental conditions to be just right. Cells of micellar-like construction take advantage of the hydrophobicity of substances with long chain hydrocarbon parts on one end and charged or polar features on the other side. Micelles are structures that spontaneously form in water. Living cells adopt a bilayer structure based upon the tendency for “likes to dissolve likes.” That is, non-polar hydrocarbon features “prefer” not to be in contact with polarized water, but rather cluster in a way that minimizes water-hydrocarbon surface contact. The effect of carbon chain structures in the biochemistry of earth is the stability of carbon-based structures and the wide variety features it can accommodate. These features include stable carbon-carbon chains as well as carbon bonds to hydrogen (H), nitrogen (N), oxygen (O) and sulfur (S) in particular. Carbon is unique in that it readily allows the formation of stable double bonds with itself or N, O, or S. Carbon also can form triple bonds with itself or N. Cyanide and acetylene are examples. The ease and stability of carbon bonded to C, N, O and S, along with the stability of multiple bonds on carbon all point to it as an excellent candidate for as the ideal building block for biomolecules.
It is often mentioned that since silicon has certain similarities to carbon why isn’t life based on it? Silicon-silicon bonds are prone to oxidation and not found in nature. Silicon is almost always found in nature as silicate in its various forms in minerals and very often in variety of silicate oligomers and polymers. Silicon-nitrogen and silicon-sulfur substances are not easy energetically. Furthermore, silicon does not form double bonds with itself or other elements. So, the variety of structural motifs silicon can form isn’t as broad as carbon. Silicon vastly prefers to be silicate in nature. Silicon is not found in biomolecules despite its high abundance in the nature.
Conclusion
I’m trying to make the point that extraterrestrial life will surely be different from life on earth at the macroscopic scale but maybe not so much at the level of molecular transformations. Every living species today trails behind it a unique evolutionary history, some of which remains in their genomes. Despite the huge variety of life forms on earth and all of the attendant structural variability that goes with it, we all share the use of DNA/RNA, proteins, carbohydrates, phosphates, lipids, calcium, magnesium, potassium, sodium, etc. All life forms on earth are able to capture and use energy as well as reproduce.
The history of life reveals an obstacle course through which organisms struggled to stay alive. Those that did survive had no way to anticipate the future and no way to prepare for it even if they were able to “anticipate” at all. The history of life is the history of challenges to survival.
Humans exist today because our ancestors going back into deep time were able to survive both anaerobic and oxygenated earth, snowball earth, competitive pressures from other life forms, vulcanism, cometary impact, solar UV radiation, chemical toxicity from the environment, disease and climate.
Today we can add stupidity to the list of survival challenges. Can we survive the results of our behavior? Humans have a brilliant streak in developing weapons- explosives, guns, nuclear, biological and chemical weapons. If all else fails, there will always be the sharp stick and club.
Humans are the way we are because of the way that natural history unfolded. A planet with the same makeup and conditions 3 billion years ago would evolve life in a different way than we went. Evolution happens because of the ability of our genetic material to be just a bit unstable and to be passed on in reproduction. But this change is a random process in both features and time. A genetic change can be fatal or helpful. The manner and schedule in which random genetic alterations happen is impossible to predict. Evolution is blind going forward. Another try at evolution is highly unlikely to produce Homo sapiens again.
Any given “intelligent” species may or may not invent or use radio technology. Therefore, they may or may not emit or receive radio transmissions. Such creatures would be undetectable using radio astronomy. Although two patents for wireless telegraphy came out in 1872, humans have only had useable wireless telegraphy since 1895 (Marconi). As of this writing, only 128 years have elapsed since Marconi sent his first long distance (1.5 mile) radio communication.
We have only had radio communications for 128 years in the entire history of our species. In order to have this invention in 1895, the European enlightenment had to happen leading to the idea of scientific inquiry and a minimum understanding of physics and chemistry. The voltaic pile had to be invented which gave way to further refinement of electricity. At minimum, the metallurgy of iron, copper and zinc (for brass) had to be in place for the for the discovery and use of electricity. The path to broadcasting and receiving radio waves required a fair degree of curiosity and industrialization.
As in the past, I will discuss some observations as a chemical scientist with cancer.
In 2013-4, I was treated for stage 4 squamous cell throat and separately, stage 4 prostate cancer and have been in remission since.
I picked up a new cancer as well as another precancer diagnosis a few weeks ago in late July, 2025. My very first colonoscopy (!!) identified several small precancerous polyps which were snipped out. The procedure was a breeze as was the much-derided colon-blow opening festivity. Propofol is amazing stuff.
Eleven days ago, I had my second partial glossectomy. The first in 2022 turned up a precancerous squamous cell lesion on the side of my tongue. The second, last week, removed a squamous cell tumor. A shallow, nickel-sized piece of tongue was removed along the middle-left edge. A skin graft from my arm was not performed, thankfully. Imagine having a hairy skin graft on your tongue!!
Prior to the surgery, I had to sanitize my arms, legs and torso with chlorhexidine (below as the gluconate), a common antiseptic. They even reamed out my nostrils with Povidone-Iodine. Incidentally Betadine is a trade name of Povidone-Iodine. First time for body-wide sanitization.
Graphic from chemical supplier TCI from Google images. This structure is the digluconate salt. Notice that the lower structure is a carboxylic acid and the chlorhexidine structure above is an is called a bibisquanide. Altogether there are 2 acid protons and 10 basic nitrogen atoms. The combination is actually an ammonium/iminium salt for water solubility.
Povidone is the polymer poly(N-vinylpyrrolidone). It is used in many medicaments and is regarded as relatively safe. The chemical structure is shown below.
Image from Wikipedia. Povidone Iodine is a broad spectrum bactericide useful against bacteria, protozoans, fungi and viruses. It is prepared by combining PVP with hydrogen iodide and iodine. It slowly releases iodine in situ.
/*begin anecdote*/
Of interest to me is the use of N-vinylpyrrolidone. In a previous life I had prepared poly-NVP by solution polymerization on many occasions as a base for experimental liquid ink charge carriers in xerographic imaging. Very simple to make. The point was to replace existing liquid inks that used flammable hydrocarbon solvents. The startup who recruited my small startup went under because the solvent they were banking on didn’t dry fast enough for their economic model. The whole thing rode on the use of low viscosity 0.5 centistoke silicon fluids.
The business plan was to provide the photocopiers at low cost and then rack in the profits on consumables- a common strategy in the printing business. The founders were all retired from the giants of the photocopier industry. They knew all about the technology except for this seemingly small ink modification.
Alas, the drying rate was far too low and the image transfer was of persistent low quality. The elderly and retired engineer behind this invention fell over dead in the middle of it. He provided the patents but never actually built a prototype or even physically investigated the suitability of silicone fluids and ink composition. It was a big handwaving exercise that the founders bought hook, line and sinker. In the end, the sinker took them to the bottom still grasping for that golden ring they so desired.
/*end anecdote*/
The ever-popular opioid fentanyl was part of the basket of anesthetics used in the partial glossectomy procedure. A little mentioned side effect of fentanyl is extreme itchiness, particularly of the face. In post-op I had this in spades and it was very uncomfortable for several hours. My interest in the chemistry of fentanyl had never fully ballooned to include side effects.
The tumor board at the university hospital I go to voted that I should undergo exploratory surgery to examine the many nearby neck lymph nodes for evidence of spread. This would point to further treatment. My throat cancer was discovered when a swollen sentinel lymph node fused to my carotid artery and decorated my neckline.
I’ll admit that a salad of pessimism and resignation with breadsticks of nihilism has arrived at my table at life’s Olive Garden. Much depends on how the upcoming lymph node surgery will come out. We’ll have to wait and see.
Assigning the stereochemical configuration of a cyclophane or a metallocene is a rare task out there for most chemists. Two classes of molecules, cyclophanes and metallocenes, have flat features that can be tough to assign priority numbers to.
I ran into an organic chemistry resource on LinkedIn that was worth zooming in on. It is a blog called MakingMolecules and it features graphics that give instruction and illustrate most aspects of sophomore organic chemistry. Having taught organic chemistry I know that nomenclature is a favorite topic among students (wink wink, nod nod), especially where stereochemical configurations are concerned. Ah …, if only the world had only chiral acyclic hydrocarbons to name. As we know, there is much, much more than that.
Finding a chiral carbon atom on most simple molecules isn’t that hard. Find a carbon atom with 4 different groups attached and then check for symmetry around it from every direction while you rotate the parts.
If it has rotational symmetry or a plane of symmetry including the atom of interest, then it may not be a chiral “center”. Molecules with a C2 symmetry axis but without a mirror plane can be chiral.
The more difficult molecules to characterize as chiral are those that have unusual rules necessary for an R or S configuration.
It is amazing what you’ll find by just looking around. While reviewing recent blood test results it occurred to me that I didn’t know the first thing about albumin as a protein. A Google word search led to numerous links but provided many images as well. The crystal structure is below.
Source. The crystal structure of human albumin. The albumin was crystallized in the presence of excess palmitic acid for x-ray analysis. Front. Immunol., 25 January 2015, Sec. Vaccines and Molecular Therapeutics Volume 5 – 2014 | https://doi.org/10.3389/fimmu.2014.00682
It is not uncommon to describe the enzyme-substrate complex as a highly specific lock and key structure. In the earlier literature is was axiomatic that enzymes are described as being highly substrate specific and use a single binding site for a given substrate. This notion is not always correct as the above graphic shows. Albumin is produced in the liver and is sort of a molecular ox cart- it can transport many substrates in the blood.
The job of human albumin is to get various substrates mobilized in the bloodstream and offer them at a desirable location. With the high molecular weight of enzymes, and the consequent low molarity available, it is astonishing that the heat of binding of substrate to enzyme can be measured at all.
One way to determine binding enthalpy and stoichiometry of a substrate to enzyme is ITC- Isothermal Titration Calorimetry. These calorimeters are available from several manufacturers such as TA Instruments and Malvern. ITC is just a type of reaction calorimeter that allows for immediate access to the reaction mixture. It is a microscale RC1 in effect. An enzyme solution can be titrated with substrate allowing for a visual determination of an equivalence point where 1 eq of enzyme active sights just matches the titrant equivalents. From such an experiment both enthalpy and stoichiometry can be measured. The image below is from TA Instruments and nicely shows the graphic output of an ITC experiment.
Source: TA Instruments product brochure. Each peak is an aliquot of titrant. Note how cleanly the signal goes to baseline between aliquots.
This graphic shows the baseline signals from titrating directly into buffer. This is subtracted from an actual run. From TA Instruments sales brochure.
Above, the background signal from the buffer represents noise in the enthalpy signal.
TA Instruments also offer equipment for so-called nano scale experiments. See below.
The TA ITC specification table. Note the minimum heat in the low volume column: 0.04 to 0.05 microJoules with a 190-microliter sample cell size.
Albumin is endowed with binding sites open to a variety of substrates. It is like a wheelbarrow or an ox cart. It can ‘carry’ numerous substrates across several categories.
The downside of such low specificity is that albumin can bind many drug compounds at the expense of dose delivery to the desired site. Doses of drugs must be adjusted to account for drug lost to blood proteins like albumin.


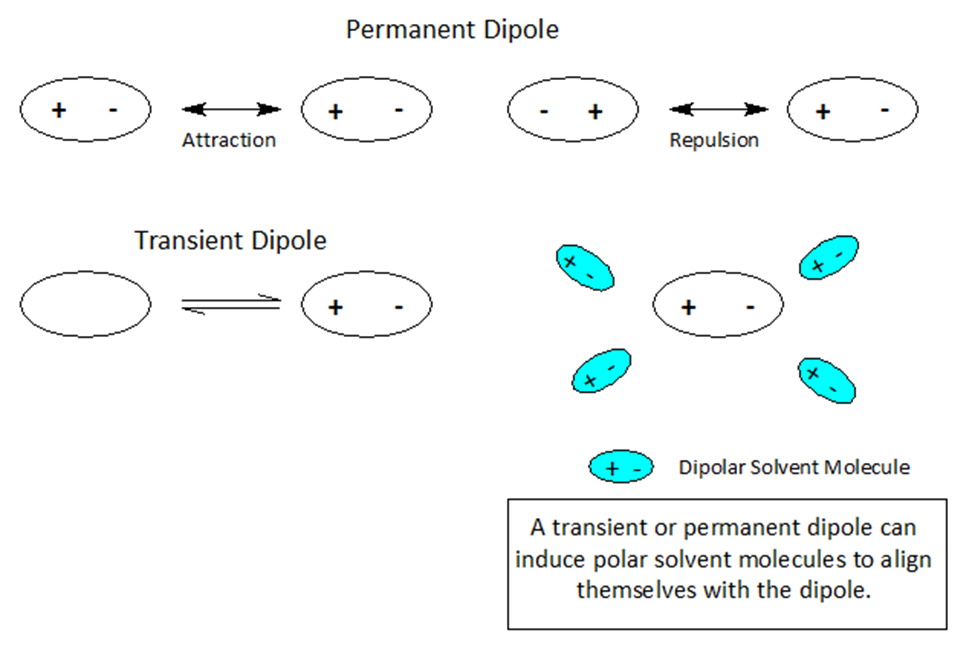
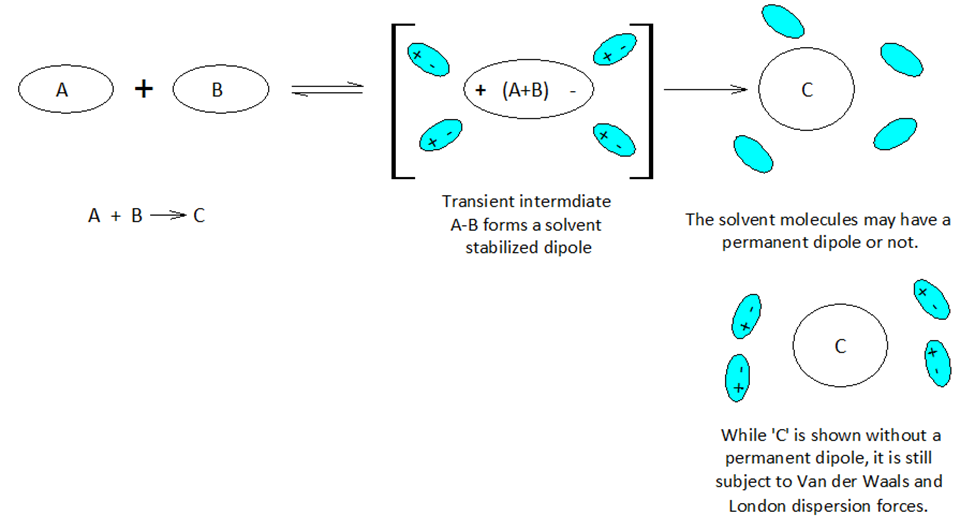
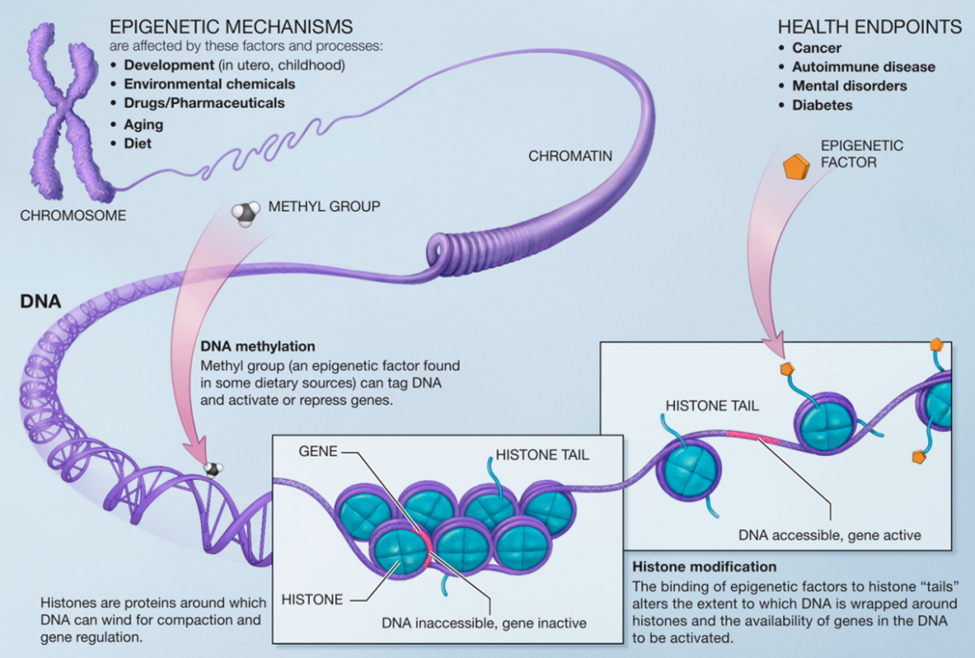


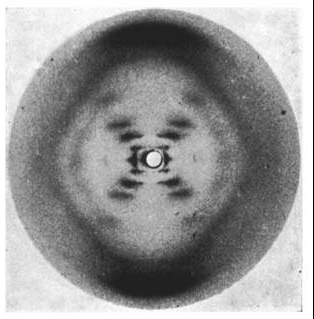
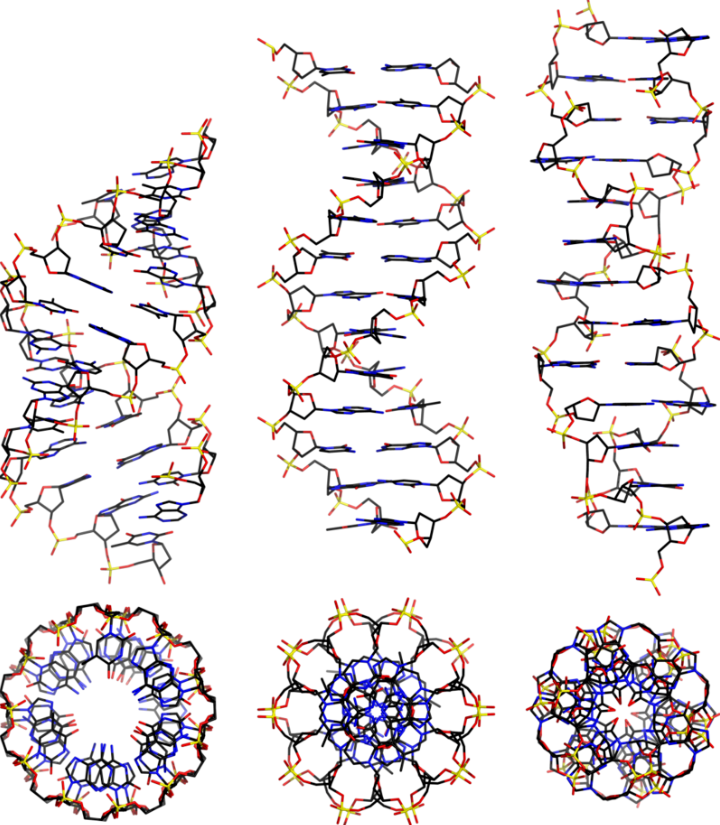
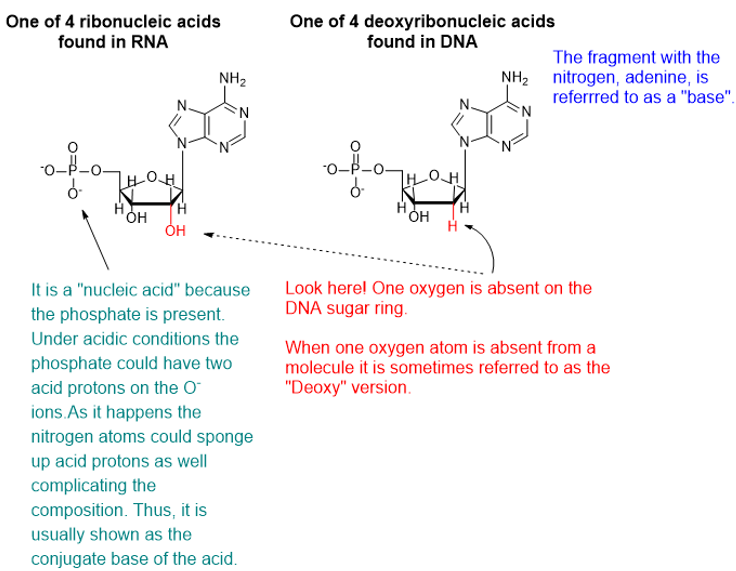
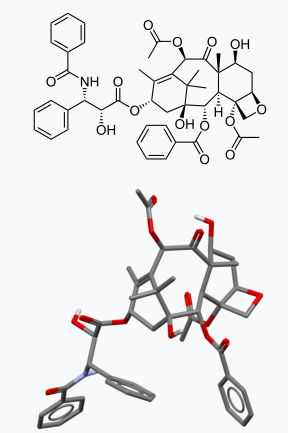

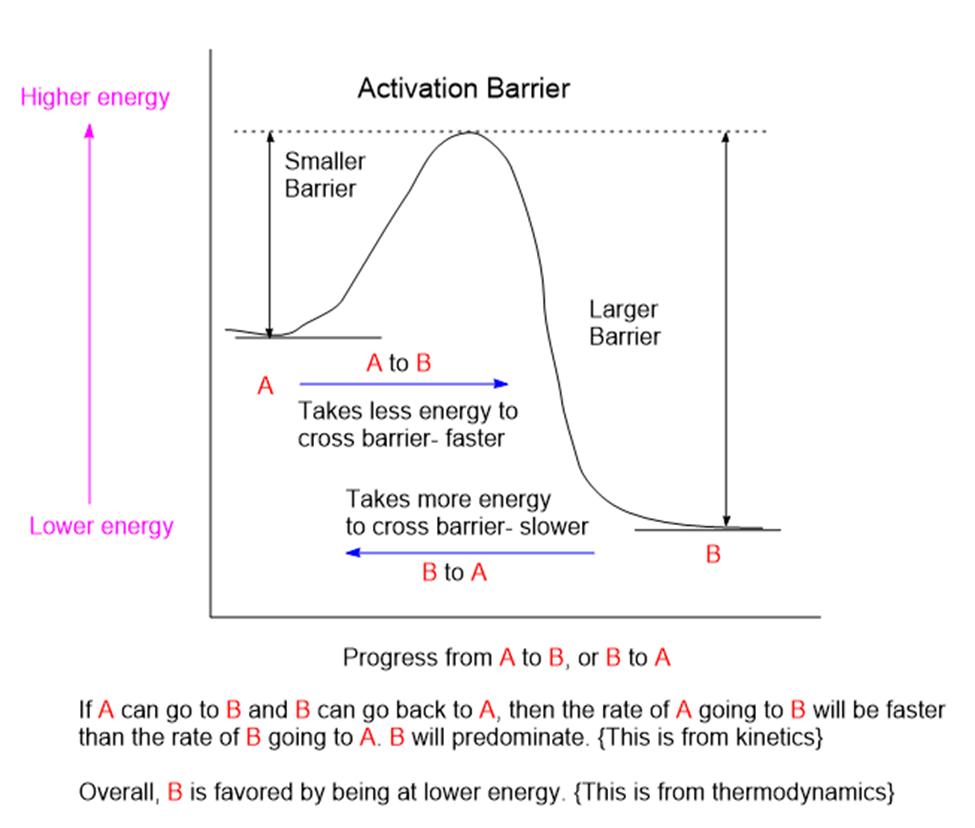
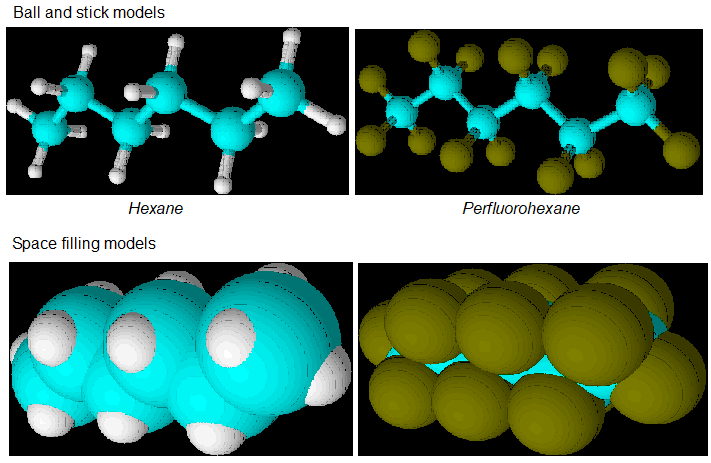
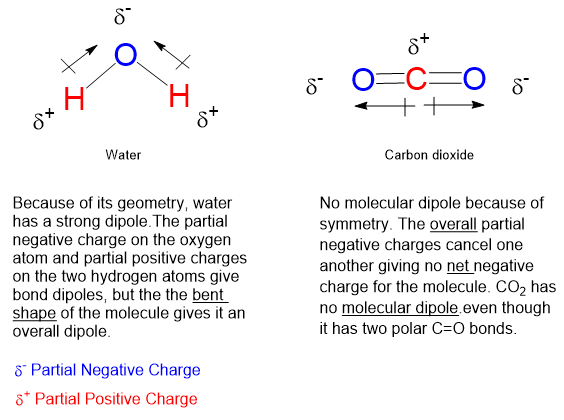


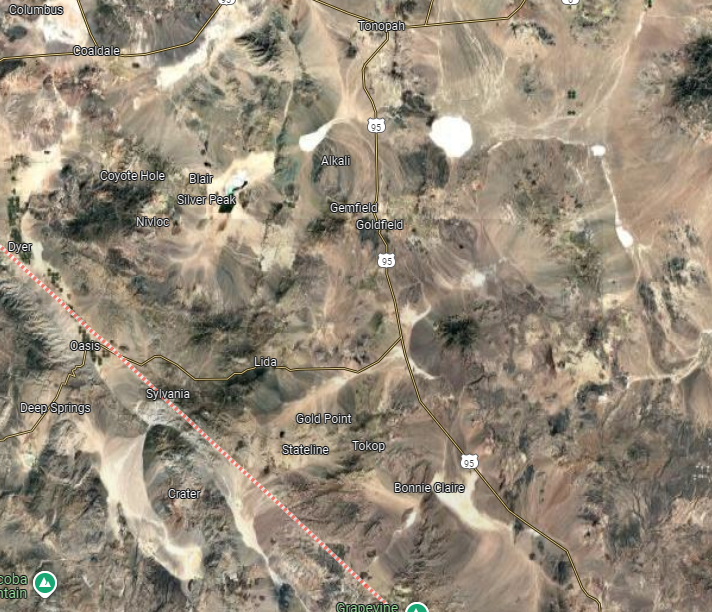
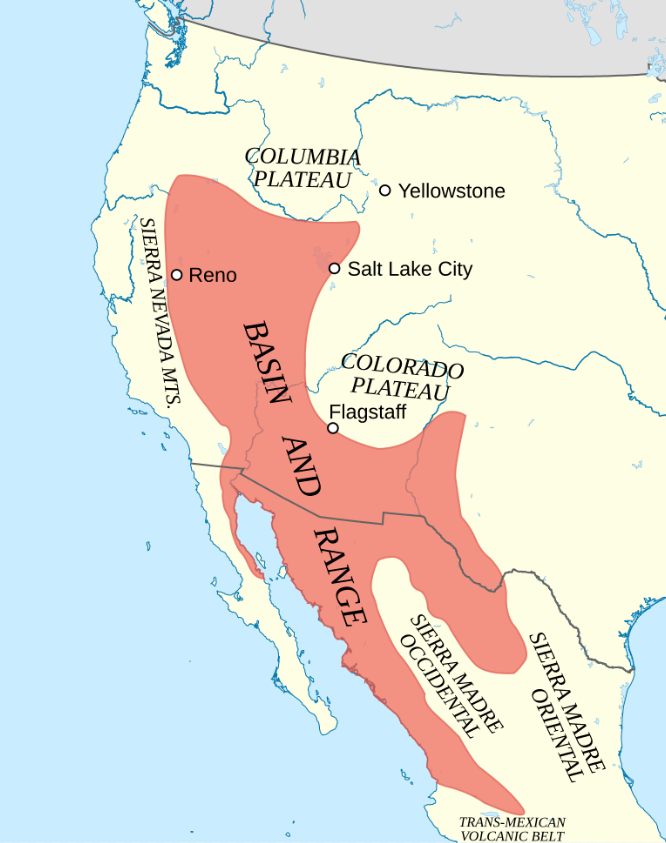

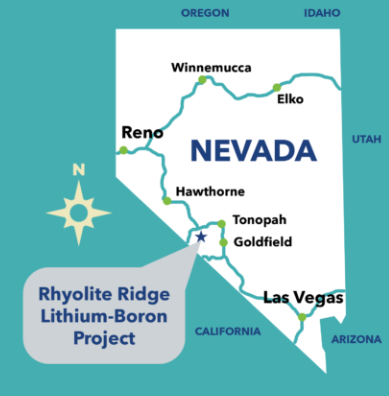


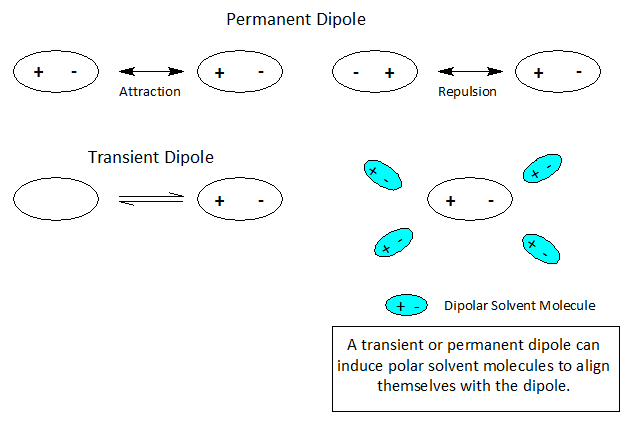{kind=link}
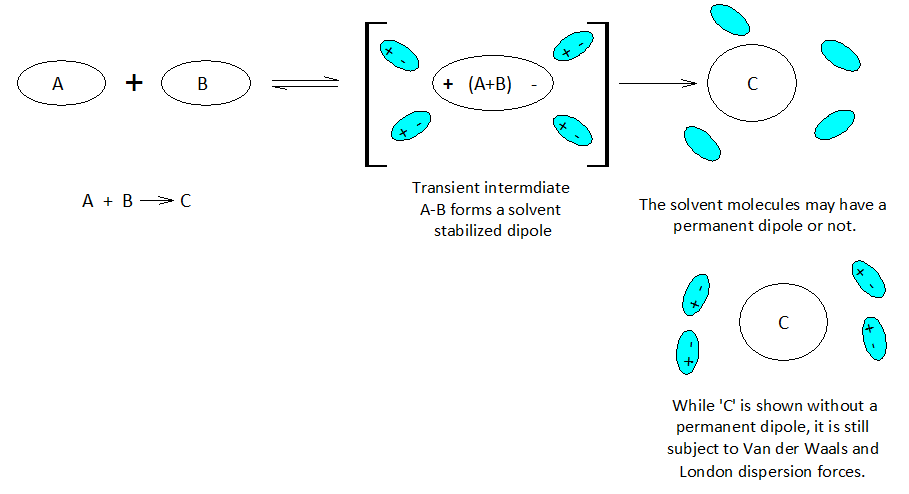{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
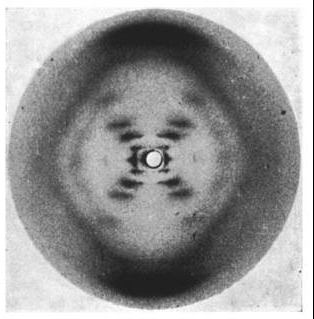{kind=link}
{kind=link}
{kind=link}
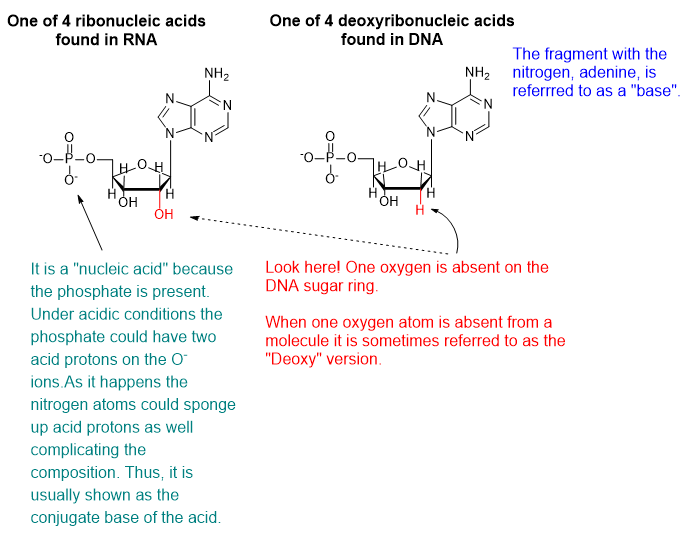{kind=link}
{kind=link}
{kind=link}