Yet another reprint of posts from the past, this time from April 11, 2008.
As usual, Th’ Gaussling’s most interesting observations of the ACS meeting are of a proprietary nature and will have to go with me to the grave. Our student and academician friends can expound openly on what lights their fires. The lusty satisfaction of compelling oratory in the darkened halls of convention centers is part of the reward for the cardinals of the academy. Members of the merchant class have to be satisfied with better dining.
People who are involved in personnel issues often speak of an employees “deliverables” as their work product. For those lucky enough to be in the academy, the work product includes teaching young minds, conducting research, and participating in the dissemination of the results in the form of papers and conferences.
For we chemists who did the deal with the devil in exchange for filthy lucre, our performance is rated somewhat differently. Like academics, our performance metric only starts with some understanding of science. Once it is possible to begin understanding a thing, the task of transforming a process or material property into an item of commerce begins. In the chemical industry we do the most important reaction of all- the transformation of chemicals into money.
The part of the brain that sees a stick on the forest floor that resembles a tool is the same part of the brain that scans a molecule and sees latent functionality or value. The extraction of value from a composition or a process is a complex anthropological activity. Product development is anthropological because it involves the use of tools and organizational structure to provide products or services that are exchanged between groups.
An industrial science group has to isolate value in some material property and contrive to bring some product or service into being. But to get it to market, the science tribe has to cooperate with those with other skills. Organizations often resemble a confederation of tribes who cooperate with complex rituals and methods of exchange.
According to BloombergNEF, the United States is on course to double its natural gas liquefaction (LNG) capacity by 2027. US export capacity is expected to rise to 169 million metric tons per year with the opening of 3 new projects slated for funding approval this year. They are- Venture Global’s Plaquemines LNG, Sempra’s Port Arthur LNG, and NextDecade’s Rio Grande LNG. This new capacity will place the US well ahead of Qatar in annual production.
Appendix–
LNG should not be confused with LPG, Liquified Petroleum Gas. LPG is a mixture of the somewhat heavier hydrocarbons propane, propylene, butylene, isobutane and n-butane. LPG is a fuel gas and can be used as an aerosol propellant and refrigerant.
LNG is composed mainly of methane (CH4) with a smaller amount of ethane (C2H6). Lesser amounts of propane and butane are isolated and sent to a separate stream. Natural gas is “sweetened” prior to cooling to remove corrosive hydrogen sulfide (H2S), carbon dioxide (CO2) gases as well as helium, mud, water, oil and mercury. Once the impurities are removed, the remaining methane/ethane mixture is cooled to −162 °C for bulk transport. On arrival at its destination, it must undergo a regasification process. In some locations seawater can be used to vaporize the LNG for injection into pipelines.
As an alternative to sea water heat transfer for regasification, LNG can be utilized for its “cold energy” potential. One application uses low temperature LNG as a refrigeration coolant for producing liquid oxygen and liquid nitrogen. Another use of the cold energy is to cool the exhaust of a gas turbine in a closed joule cycle with argon as the fluid.
Since we are talking about gaseous hydrocarbons, there is also a category of liquid hydrocarbons called condensates that accompany the production of natural gas and must be channeled into a separate processing stream because, well, they are liquid. Raw natural gas straight out of the ground may have varying amounts of condensates-
Crude oil wells can produce natural gas called associated gas and condensates may be entrained in the gas flow.
Dry gas wells produce gas that have no associated liquids.
Condensate wells produce natural gas with associate natural gas liquid.
Wikipedia explains the condensate situation in greater detail.
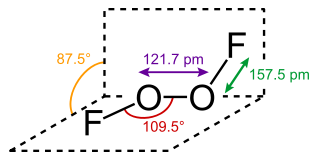
Just today, as the open door to my golden years stands gaping before me, I learned of a substance called dioxygen difluoride, FOOF. It’s also known as perfluoroperoxide at the NIST Chemistry WebBook site. Seems like I’m always the last one to the oxidizer party. This rather unhappy substance can be prepared as shown below. The word is that the orange-yellow solid is only stable below −160 °C.
Image from Wikipedia. https://en.wikipedia.org/wiki/Dioxygen_difluoride. A mixture of fluorine and oxygen gas were heated to 700° C then, according to the abstract “rapidly cooled on the outer surface of stainless steel tubes. The tubes were refrigerated by a liquid oxygen bath pressurized to >7600 torr with helium. Six grams of O2F2 were produced in less than an hour.”
Derek Lowe mentioned in one post in his Blog In the Pipeline that FOOF was in the list of materials he won’t work with. Derek also mentioned chlorine trifluoride. A method of preparing this substance is shown below. This substance is a powerful fluorinating agent and reacts in hypergolic fashion with asbestos and sand according to chemist John Drury Clark. Clark wrote a book called Ignition! An Informal History of Liquid Rocket Propellants based on his experiences with rocket propellant research. Clark said that the great toxicity of ClF3 was the “least of its problems”. It’s ability to react in a hypergolic manner with nearly everything was a barrier to its use. It could be stored in metal containers that were first passivated with fluorine gas.
3 F2 + Cl2 → 2 ClF3
Uranium hexafluoride is produced with chloride trifluoride-
U + 3 ClF3 → UF6 + 3 ClF
According to Wikipedia, ClF3 is used to clean Chemical Vapor Deposition (CVD) chambers. Not surprisingly, prior to WWII the Nazis had experimented with ClF3 as a chemical warfare agent called N-Stoff. Production halted when the Red Army overran the facility in 1945. The substance was never used in war.
This post is an update of a post I wrote on Mole-Day of 2011. It is a brain dump that summarizes much of what I’ve learned about dealing with potentially explosive chemicals in the manufacturing environment. Very few chemists actually have to deal with explosive chemicals in their work activities. It is actually quite uncommon. No doubt some important considerations have been left out and for that I apologize.
The Prime Directive: If you choose to bring in or make a chemical substance in your facility, you must develop in-house expertise in the safe handling and use of that substance. Do not expect to rely on outside expertise for it’s safe use. Always strive to build in-house expertise in regard to chemical properties and safety- never farm this out to consultants. This includes proper engineering and broad knowledge of reactive chemical hazards.
Safety has a substantial psychological component. You can build into a chemical manufacturing process extensive engineering and administrative controls for safe operation. These layers of control are concrete and definable. What is fuzzy, however, is the matter of how people behave. In particular, I’m thinking of getting people to behave in a particular way over the long haul. Keeping people operating safely over long periods of time where no adverse events happen poses special problems. Especially in regard to low frequency, high consequence events. Cutting corners and improper use of PPE is not uncommon and should be expected. Something expected can be watched for continuously.
In safety training I mention that handling a hazardous material is like handling a rattle snake. You have to exercise the due caution every single time you pick up that snake. You do not accumulate and bank safety credits for previous safe handling. Everybody understands this already at some level. But the possibility of drift in safety practice over time needs to be emphasized.
The best strategy I know of besides complete process automation is recurrent safety training along with vigilant management. Successful safety management requires proper supervision by alert supervisors. Management by walking around helps with this. Well written process instructions that anticipate practical problems are essential. Holding people accountable for following Standard Operating Procedures is critical. Working conditions conducive to focus are always good. Operational rotation with may be helpful.
In chemical safety, the biggest worry is typically the potential for an explosion. What should you do if a raw material or product in a process may be explosive or has explosive features on the molecule? Good question. First, someone in the R&D chain of command should have knowledge of the list of known explosophores. It’s not a big list. PhD chemists in R&D should know this anyway. Explosive molecules have certain chemical bonds that are weakest and are known as “trigger bonds“. It is thought that the rupture of these trigger bonds initiates explosive decomposition of the substance.
Just because a material has explosive properties does not automatically disqualify it for use. Azides and nitro compounds are used safely every day. But, to use a chemical safely you must accumulate some knowledge on the type and magnitude of stimulus that is required to give a hazardous release of energy.
For any given hazard, it is my personal policy to learn as much about the nature of the hazard at the chemical and bulk level as I can. I believe that it is important to know more about something than what is immediately called for. That is the difference between education and training. This is how you build expertise.
Some comments on the release of hazardous energy. Hazardous energy is that energy which, if released in an uncontrolled way, can result in harm to people or equipment. This energy may be stored in a compressed spring, a tank of compressed gas, the stable chemical bonds of a flammable material, the unstable chemical bonds of an explosive material, or as an explosive mixture of air and fuel. A good old fashioned pool fire is a release of hazardous energy as well. Radiant energy heating from a pool fire can easily and rapidly accelerate nearby materials past the ignition point. Good housekeeping goes a long way towards preventing the spread of fires.
Applying and accumulating energy in large quantities is common and actually necessary in many process activities. In chemical processing, heat energy may be applied to chemical reactions. Commonly, heat is released from chemical reactions at some level ranging from minimal to large. The rate of heat evolution in common chemical reactions can be simply and reliably managed by controlling the temperature or rate of addition of reactants where two reactants are necessary. However, reactions do not always evolve significant power output immediately on mixing of the reactants.
Induction periods are potentially dangerous and must be identified prior to scale up. The appearance of an exotherm very early in a feed operation is a good indication that the reaction has begun. However, a thermogram from a reaction calorimeter showing the temperature and power output (watts) versus the feed mass will indicate if the reaction is slow and accumulation of reagent (energy) is occurring. This can be teased out early by adding a small shot of reactant feed (a few %) and watching the power profile. The ideal profile is where the power output starts promptly, peaks and then promptly decays to baseline. This is a good indicator of the absence of accumulation. Generally, the kinetics are most favorable at the beginning of the reagent feed and taper off to zero as reactants are consumed. Some accumulation is usually tolerable from the heat load perspective. This is a good thing because a thermogram showing some accumulation could lead to an unnecessarily long feed time. A reaction calorimeter can give the peak wattage per kilogram of reaction mass. An engineer should be able to estimate the maximum controllable heat flux for a given reactor. Without being too specific, it is in the range of several tens of watts per kg of reaction mass according to one reference I know.
There are explosive materials and there are explosive conditions. If one places the components of the fire triangle into a confined space, what may have been simple flammability in open air is now the makings of an explosion. Explosive materials have two legs of the fire triangle built into the molecule- the oxidizer and the fuel separated by only nanometers. However, the composition of the explosive itself may not produce a balanced reduction/oxidation reaction. The oxygen balance is a easily calculated number that will indicate whether or not there is an excess or deficit of oxygen in an explosive substance. For example, ammonium nitrate has a 20 % excess of oxygen. Fuel oil can be added to bring the fuel/oxidizer ratio into redox balance. This mixture is referred to as ANFO.
In a chemical explosion, heat and increasing pressure can do PV work on the contents and containment. Minimally, the outcome will be an overpressure with perhaps the blowing of a rupture disk on a reactor. In another situation, the equipment may blow apart and send fragments flying away at high speed with an expanding fireball.
There is a particular type of explosive behavior called detonation. Detonation is a variety of explosive behavior that is characterized by the generation and propagation of a high velocity shock through a material. A shock is a high velocity compression wave which begins at the point of initiation and propagates throughout the bulk mass of explosive material. Interestingly, because it is a wave, it can be manipulated somewhat by reflection and refraction. This is the basis for explosive lensing and shaped charges. It is characteristic of detonations to produce shredded metal components. Detonations have a very large rate of pressure rise, dP/dt. The magnitude of dust explosions is commonly performed by a few commercial test labs out there. One of the important test results is the Kst value showing the magnitude of the explosive force.
Detonable materials may be subject to geometry constraints that limit the propagation of the shock. A cylinder of explosive material may or may not propagate a detonation wave depending on the diameter. Some materials are relatively insensitive to the shape and thickness. A film of nitroglycerin will easily propagate as will a slender filling of PETN in detonation cord. But these compounds are for munitions makers, not custom or fine chemical manufacturers. The point is that explosability and detonability is rather more complex than one might realize. Therefore, it is important to do a variety of tests on a material suspected of explosability. The type and magnitude of stimulus necessary to produce an explosion must be understood for safe handling and shipping.
A characteristic of detonable explosives is the ability to propagate a shock through the bulk of the explosive material. However, this ability may depend upon the geometry of the material, the shock velocity, and the purity of the explosive itself. There are other parameters as well. Marginally detonable materials may lose critical energy if the shape of the charge provides enough surface area for loss of energy.
Explosive substances have functional groups that are the locus of their explosibility. A functional group related to the initiation of explosive behavior, called an explosophore, is needed to give a molecule explosability. Obvious explosophores include azide, nitro, nitroesters, nitrate salts, perchlorates, fulminates, diazo compounds, peroxides, picrates and styphnates, and certain hydrazine moieties. Other explosophores include the hydroxylamino group. HOBt, a triazole analog of hydroxyamine, hydroxybenzotriazole, has injured people, destroyed reactors and caused serious damage to facilities. Anhydrous hydroxylamine has been the source of a few plant explosions as well. It is possible to run a process for years and never cross the line to runaway as was the case for these substances.
Let’s go back to the original question of this essay. What do you do if you find that a raw material or a product is explosive? The first thing to do is collect all available information on the properties of the substance. In a business organization, upper management must be engaged immediately since the handling of such materials involves the assumption of risk profiles beyond that expected.
At this point, an evaluation must be made in relation to the value of the product in your business model vs the magnitude of the risk. Dow’s Fire and Explosion Index is one place to start. This methodology attempts to quantify and weight the risks of a particular scenario. A range of numbers are possible and a ranking of risk magnitude can be obtained therein. It is then possible to compare the risk ranking to a risk policy schedule generated beforehand by management. The intent is to quantify the risk against a scale already settled upon for easier decision making. A problem with this approach is that it requires numerical values for risk which might be difficult to come by.
But even before such a risk ranking can be made, it is necessary to understand the type and magnitude of stimulus needed to elicit a release of hazardous energy. A good place to start is with a DSC thermogram and a TGA profile. These are easy and relatively inexpensive. A DSC thermogram will indicate onset temperature at a given temperature ramp rate and energy release data as a first pass. Low onset temperature and high energy release is least desirable. High onset temperature and/or low exothermicity is most desirable.
What is more difficult to come to a decision point on is the scenario where there is relatively high temperature onset and high exothermicity. Inevitably, the argument will be made that operating temperatures will be far below the onset temp and that a hazardous condition may be avoided by simply putting controls on processing temperatures. While there is some value to this, here is where we find that simple DSC data alone may be inadequate for validating safe operating conditions.
Onset temperatures are not inherent physical properties. Onset temperatures are kinetic epiphenomena that are dependent on the sensitivity of the instrument, sample quality, the Cp of both the sample and the crucible, and the rate of temperature rise. What may be needed once an indication of high energy release is indicated by the DSC is a determination of time to maximum rate (TMS). While this can be done with special techniques in the DSC (i.e., AKTS), TMR data may be calculated from 4 DSC scans at different rates, or it may be determined from Accelerated Rate Calorimetry, or ARC testing. Arc testing gives time, temp, and pressure profiles that DSC cannot give. ARC also gives an indication of non-classical liquid/vapour behavior that is useful. ARC testing can indicate the generation of non-condensable gases in the decomposition profile which is good to know.
Time to maximum rate is measured in time at a specified temperature. Many people consider that a TMR of 24 hours at the process temperature is a minimum threshold for operational safety. Others might advise 24 hours 50 or 100 C above the maximum operational temperature. If you contemplate using this parameter, it is critical to get testing from a professional lab for a time at a particular temperature. This kind of test will produce a formula that you can calculate TMR values at a given temperature. Bear in mind, however, that no outside safety consultant will tell you what you must do for liability reasons. You must develop enough in-house expertise to make this decision for yourself.
The standard tiered test protocol for DOT classification is a good place to start for acquiring data on explosive properties. Several companies do this testing and give ratings. There are levels of testing applied based on the result of what the lower series tests show. Series 1 and 2 are minimally what can be done to flesh out the effects of basic stimuli. What you get from the results of Series 1, 2, and 3 are a general indication of explosibilty and detonability, as well as sensitivity to impact and friction. In addition, tests for sensitivity to electric discharge and dust explosion parameters should be performed as well.
The card gap test, Konen test, and time-pressure test will give a good picture of explosive behavior. The Konen test indicates whether or not extreme heating can cause an explosion sufficient to fragment a container with a small hole in it.
BOM or BAM impact testing will indicate sensitivity to impact stimulus. Friction testing gives threshold data for friction sensitivity.
ESD sensitivity testing gives threshold data for visible effects of static discharge on the test material. Positive results include discoloration, smoking, flame, explosive report, etc.
Once the data is in hand, it is necessary to sift through it and make some business decisions. There is rarely a clear line on the ground to indicate what to do unless there is already a policy on decision making here. What testing results will indicate is what kind of stimulus is necessary to give a positive result with a particular test. It is up to your in-house experts and management to decide the likelihood of exposing the material to a particular stimulus. Will it be possible to engineer away the risk or diminish it to an acceptable level? The real question for the company is whether or not the risk of processing with the material is worth the reward. Everyone will have an opinion.
The key activity is to consider where in the process an unsafe stimulus may be applied to the material. If it is thermally sensitive in the range of heating utilities, then layers of protection guarding against overheating must be put in place. Layers of protection should include multiple engineering and administrative layers. Every layer is like a piece of Swiss cheese. The idea is to prevent the holes in the cheese from aligning.
If the material is impact or friction sensitive, then measures to guard against these stimuli must be put in place. For solids handling, this can be problematic. It might be that preparing the material as a solution is needed for minimum solids handling.
If the material is detonable, then all forms of stimulus must be guarded against unless you have specific knowledge that indicates otherwise. Furthermore, a safety study on storage should be performed. Segregation of explosable or detonable materials in storage will work towards decoupling of energy transfer during an incident. By segregating such materials, it is possible to minimize the adverse effects of fire and explosion to the rest of the facility.
With explosive materials, electrostatic safety is very important. All handling of explosable solids should involve provisions for the suppression of electrostatic charge generation and accumulation. A discharge of static energy in bulk solid material is a good way to initiate runaway decomposition of an energetic material. Unfortunately, some explosive substances may not require the oxygen leg of the fire triangle so, in this case, inerting with nitrogen won’t be preventative.
Safe practices involving energetic materials require an understanding the cause and effect of stimulus on the materials themselves. This is of necessity a data and knowledge driven activity. Handwaving arguments should also be suppressed in favor of data-driven analysis.
It was announced that a US company will be supplying critical components for Electric Vehicle (EV) batteries to Panasonic. Redwood Materials, Inc., is set to supply EV battery cathode components from its facility in Kansas City. Redwood Materials was founded to close the battery recycle loop by JB Straubel. Straubel was a co-founder and former CTO of Tesla.
A lithium-ion battery doesn’t just rely on lithium. Other substances work together with lithium and the whole composition will vary between manufacturers. The Wikipedia entry for lithium-ion batteries lists the Panasonic cathode material as LiNiCoAlO2. Panasonic works in cooperation with Tesla to supply batteries using Lithium Nickel Cobalt Aluminum Oxide cathode batteries. As alluded to above, Redwood will be supplying cathodes made of recycled battery materials.
The lithium battery electrolyte is almost always contains a lithium salt such as LiPF6, lithium hexafluorophosphate, in a non-aqueous organic carbonate electrolyte like ethylene or propylene carbonate. These two carbonates function as high boiling, polar aprotic dispersants. The substances are cyclic carbonate ester compounds and have a high dielectric constant. The high dielectric constant means that the molecules are polar enough to coordinate Li+ ions to aid in electrolyte mobilization of the Li salt. The electrolyte may also contain a solvent like diethyl carbonate to decrease viscosity and lower the melting point. The PF6 anion is a large, charge diffuse, weakly coordinating anion that helps keep the lithium cation mobilized and loosely bound in the polar aprotic carbonate solution. This anion is inert enough and lends solubility in organic solvents making it useful for many applications. Ammonium salts with PF6 anion are often used as ionic liquids. Weakly coordinating anions are used to allow the corresponding cation to be partially unsolvated and therefore more available for reaction chemistry.
Both in producing power and in recharge, when electrons are being passed around between chemical species and changing oxidation states, it means that chemical changes are occurring. When chemical changes (reactions) are happening, it means that heat is being absorbed or evolved. In the emission of heat, the amount of heat energy per second (power) produced can be large or small. It is critical that the temperature of the battery not exceed the boiling point of the lowest boiling component which may be the carbonate dispersant, as in ethylene carbonate (bp 243 C) or viscosity modifier like diethyl carbonate (bp 126 C). A liquid phase internal to the battery flashing to vapor can overpressure the casing and rupture the battery. A liquid changing into a vapor phase wants to increase its volume by from ~650 to 900 times or beyond. To make matters worse, a chemical reaction generally doubles its rate with every 10 degrees C of temperature rise. Runaway reactions generate runaway heat production.
Lithium batteries have flammable components such as ethylene carbonate (flash point 150 C) and diethyl carbonate (flash point 33 C) that could be discharged and ignited if the battery bursts open, possibly leading to ignition of the surroundings, be it in your pants pocket or in the cargo hold of a passenger aircraft.
This article amounts to a plea to analytical chemists, supervisors, and organizations who use perchloric acid to make the effort to understand its reaction chemistry, as an acid or salt, and the peculiarities of the numerous mixtures used in analytical sample digestion. If your organization uses standard methods of digestion via one of the many acid mixtures and temperatures, it behooves your organization to have at least one individual on site who understands a bit more than just the procedure. If there is an incident of some kind involving perchloric acid, be it a spill, splash, or worse, having a grasp of the real hazard presented before you is useful. It is possible to underreact or overreact to any given incident scenario.
I am not an analyst. My interest is to understand reactive chemical hazards and devise means for preventing the transition from hazard to danger. Whether someone uses perchloric acid or not makes no difference to me. I have no investment in perchloric acid. However, I’m greatly interested in users being informed.
Comments on Safety Training
Safety training is commonly executed as a result of company policy where documentation of satisfactory completion is collected and filed. For lab chemists this includes training sessions on chemical storage, fire safety, fire extinguisher training, hazardous waste practices and regulations, storm water regulations, company safety and health SOP training, building evacuation, general lab safety, and perhaps basic first aid.
Often safety training sessions are canned professional video presentations or a corporate home brew of PowerPoint slide shows followed by some Q&A and a quiz. It is what I refer to as infotainment. Attendees may watch a video with dramatized incidents while the voiceover describes what should have happened. This approach is not without merit or some success, but this passive approach may not be of lasting value. Furthermore, it is a very sketchy assumption that such passive training will result in proper decision making in an off-normal circumstance where hazard may transition to danger.
The military has solved this problem long ago by mastering the art of the drill. They realize that if you need people to respond in a particular way rapidly, they have to be trained and drilled. In times of peace, the military has the opportunity to train and drill to maintain operational readiness. This is one way to address the difficult problem of low probability, high consequence scenarios. Industry as a whole, however, may not inclined to offer a lot of free time to dedicate to training. Man-hours in drills subtract from productivity. In my opinion, much of industrial management suffers from a lack of imagination in this matter. Safety training and drills are cost overhead. But, what you lack in training hours may be made up for by effective mentoring.
We live in the age of OSHA regulations. Of importance to the process industry is Process Safety Management or PSM. The mission of OSHA is copied and pasted below.
The Wikipedia link below gives an excellent summary of OSHA regulations relating to the chemical process industry. PSM in 29 CFR §1910.119 titled Process safety management of highly hazardous chemicals, is a regulatory framework covering all aspects of safety management and threshold quantities (Appendix A) of highly hazardous materials. Whether your facility is operating at the PSM scale of operation or not, employers have a duty to assure a safe operating environment for their employees. In my view, PSM regulations frame a safety mindset and diligence that is useful outside of PSM reach. Given that a debilitating injury, fatality, explosion or major fire will bring the unblinking eye of regulators and possible litigation, sensible practices found in 29 CFR §1910.119 that are woven into your chemical safety SOPs are in the direction of goodness. Again, this is my view and should not be construed as legal advice. Your chemical safety plan is your responsibility alone.
Finally, a word to lab managers and supervisors. I cannot point to a ancient stone or a law of nature that commands that leaders be effective instructors and mentors. But I can throw an idea on the table which is that as a senior employee in a supervisory role, you have a moral obligation to your charges to make sure that they practice their art with diligence and in a safe manner. The best way I know of is to train staff thoroughly in lab operations and have high expectations of your staff. Management by wandering around can be very effective in maintaining discipline and keeping tabs on your shop. Besides, you should be walking around and asking questions anyway.
HClO4 – The Meat and Potatoes
There is much to know about the chemistry of perchloric acid digestion beyond it’s renowned acidity and explosive potential. Appreciating the corrosivity and close adherence to standard laboratory techniques are necessary but not always enough. One such circumstance begging for informed action is method development. In researching this topic I was a little surprised to find that many important details are buried in the primary literature. Worse, a few key references are downright difficult to obtain. By important details, I mean whatever information might help define the safe operating window for a given digestion, or, better put, under what circumstances might a digestion procedure transition from hazardous to dangerous.
The major supplier of perchloric acid and perchlorate salts in the USA is GFS Chemicals in Powell, OH. The founder of this company, G. Frederick Smith was, and remains posthumously through his writings, a top authority on the properties of this acid and numerous perchlorate salts as the result of his many decades of research. Laboratory quantities of perchloric acid can be had from GFS and the usual group of research chemical suppliers.
It is easy to find MSDS data and exemplar laboratory safety guides on your browser detailing sensible storage and use policy. Several found in google-space stand out in my opinion as comprehensive perchloric acid safety documents and SOP’s; UC Berkeley; Boston University; MIT; Harvard; British Columbia Code for Mines to name a few. Again, this is my opinion- form your own. If your perchloric acid “policy” is limited to an MSDS document and perhaps a few safety statements found in a procedure, then I would urge someone in your organization to take it upon themselves to dig in a little deeper. Generate SOPs for all aspects of the perchloric acid life cycle in your facility.
There are many accounts of incidents with perchloric acid that should convince even the most refractory skeptic of the potential for a violent release of energy. There is a perchloric acid incident that stands out as an example of the dangers of a chemical ignorance. It happened February 20, 1947, when a large and violent explosion killed 17 people and led the city of Los Angeles to specifically bar the use of perchloric acid (1) through numerous sections of it’s zoning code.
The most common laboratory use of perchloric acid is in the analytical digestion of samples containing a matrix of organic matter, sludge, tissue, biomass or organic chemicals. There are a great many lab procedures to be found by an internet search including Chemical Abstracts (CAS), the AOAC Official Methods of Analysis manual, and ASTM relating to HClO4. Numerous policy and prudent practices documents can be downloaded from well established institutions that outline some very sensible policies regarding the storage, use, and disposal of HClO4. One particularly good source for sample digestion methods across the periodic table is from Inorganic Ventures. Kudos to Dr. Paul Gaines and this company for the quality of their products and their willingness to share their expertise in trace element analysis.
A search of Chemical Abstracts will turn up many research papers giving digestion procedures in the experimental section. However, it is not often made clear how the workers came upon their particular digestion conditions other than from a reference in an earlier procedure. This is because these papers are about the use and not about the chemistry of digestion. Most of the procedure writers will have done their diligence and provide warning about hazards. What may be omitted within papers that use the HClO4 procedure are the boundaries of safe operation and how the reactivity may vary with concentration and temperature.
For greater detail one must look elsewhere and well back into the 20th century. Much useful information on HClO4 and its salts is to be found in papers from the 1930’s thru the 1970’s. Because of their energetic properties, the propellant and explosives folks usually expand on energetic materials including perchlorates, and yes, they go into some great and admirable detail (2). However these sources tend to be thermochemical in nature and perhaps not a lot of immediate help to a bench chemist.
Unlike many other reagents in the laboratory, perchloric acid can have a downside with immediate negative safety consequences. In particular, if one is aiming to develop a digestion procedure for a new type of sample, say, something with a mixed organic/inorganic matrix or certain heteroatoms compounds with nitrogen or sulfur, it behooves the chemist to take a serious interest in rooting out information about the safe operating boundaries of perchloric acid and what kinds of materials may be problematic. A perchloric acid MSDS will inform you of potential safety hazards, hazard classifications, etc., but a well researched and validated procedure can go far towards keeping you out of trouble. I would recommend that at least one person at your organization be more thoroughly educated in the chemistry of perchloric acid digestion, or wet ashing as it is called. Unlike some other strong acids, contact with organics may have immediate explosive consequences. And by explosive I mean violent, deafening, shrapnel-blasting detonations. Hazardous contact can include contact of hot concentrated acid on paper, on sample material, or even contact of perchloric acid vapor on a gloved hand passing through fumes.
There are some particularly comprehensive and broadly informative publications covering perchloric acid chemistry. A more recent work by John Long (3) of GFS is particularly insightful in regard to drawing a line between perchlorate salts and perchloric acid. The 1960 publication Perchlorates: Their properties, manufacture, and uses by J.C. Schumacher (4) contains an informative chapter (Ch 11) on perchloric acid safety. Perhaps the most useful reference is a book available from GFS (5) or Amazon titled Perchloric Acid and Perchlorates, by A.A. Schilt. The 2nd edition in particular contains a great many useful references.
On heating at ambient pressure, aqueous perchloric acid will concentrate by distillation to a constant boiling azeotrope of 72.5 % HClO4 and water. At this composition its number of waters of hydration is slightly greater than two. In the climb from ca 160 °C to a bp of 203 °C at 1 atm, the 72.5 % acid will transition from being “just” a hot super acid to a super acid and a potent oxidizer.
In the gas phase, this acid can decompose via a radical pathway leading to the evolution of Cl2, O2, H2O either abruptly or after an time interval (6). Note that when something quite hot abruptly decomposes to a greater number of moles of gaseous products, there can be plenty of potential for destructive pressure effects.
For the uninitiated, HClO4 is a “super” mineral acid capable of complete dissociation in aqueous concentrations up to about 4 molar (7). The dissociated form in water is H3O+ ClO4-, or oxonium perchlorate. This is normal Brønsted acid behavior in water, but three things set this acid apart from others, even nitric acid: i) due to the extremely weak coordinating ability of the perchlorate anion, the acid proton is extraordinarily mobile and reactive; ii) at room temperature the anhydrous acid will at some point spontaneously explode; and iii) in concentrated aqueous form at elevated temperatures, say > 160 ºC, the acid becomes an increasingly potent oxidizer with temperature.
The perchlorate anion has a central chlorine atom, formally +7, that sits in a tetrahedral array of four O2- anions to make it anionic. On average the negative charge is spread over the surface of the symmetric anion making the negative charge diffuse with the enthalpy of formation unfavorable to close ion pairing. The perchlorate anion is only weakly attracted to a given cation like H3O+ or oligomers and as such, allows the H3O+ (or larger clusters) to reside in a solvent shell unencumbered by tight ion pairing, depending on the nature of the solvent. Perchlorate salts can have very high water solubility and, in the case of magnesium perchlorate, serve as an excellent desiccant. One exception to the high solubility of perchlorates is potassium perchlorate at only 1.5 g per 100 mL H2O at 25 °C.
Perchlorates: A review of their thermal decomposition and combustion, with an appendix on perchloric acid, G.S. Pearson; Rocket Propulsion Establishment; October 1968. http://www.dtic.mil/dtic/tr/fulltext/u2/857556.pdf
In this post I’ll feature a particularly well-done group of videos on precious metals prospecting, milling and smelting. The producer of this content is Jason Gaber at Mount Baker Mining and Metals, MBMM. The website says that Jason is a geophysicist. His company manufactures small-scale industrial grade equipment for the processing of ore. He produces videos that show how things are done in prospecting, mining, and even smelting. His videos give long, lingering views of the milling and smelting processes in operation. I was interested in particular in the process of cupellation, which has always been a bit of a mystery.
Gold ore is dropped into a crusher then pulverized to millimeter-size with a hammer mill. The finely divided ore is then fed onto a shaker table for separation by density with flowing water. The shaker table is a mechanical separation method that allows the isolation of metal fines without chemical processing methods. No cyanide or mercury here. The only waste materials are the pulverized ore tailings.
Editorial comment: To be sure, there is nothing innocent about ore tailings. The large surface area along with the presence of sulfides and water allow air to oxidize the sulfur to strong mineral acid and accelerate the leaching of hazardous metals into streams over the long term. It is very damaging to wildlife and municipalities that draw water from the stream and rivers. Water pollution is a problem all around the American West. Metals are forever.
The smelting videos are interesting for a chemist to watch. Jason uses his knowledge of pyrometallurgy to extract the values and partition impurities away from the target metal. Of course, chemists will recognize this as high temperature inorganic chemistry. Before watching this, I had a poor understanding of the importance of fluxes and slag. Jason quantitatively formulates custom fluxes to fit the problem as he sees it. He uses iron bars for redox processes to change the chemical composition of the melt and give a better partitioning of components.
The goal in smelting is to get a clean separation of the metal value from the ore by partitioning between liquid phases. Lead is often used as a “collector” metal to accumulate reduced metal species as a separate liquid phase on the bottom of the melt. The upper slag phase is a complex mixture of the ore matrix material and contains silicates, aluminates, and a dog’s lunch of other undesirable substances. And. not all metals are miscible or highly soluble in the collector phase, so there is some art in this.
Jason also discusses matte and how to deal with it. Matte is frequently discussed in 19th century works on gold smelting, but this was before atomic theory or sophisticated analytical chemistry. Matte was something to place in a reverberatory furnace and calcine. Sulfides in the matte were converted to oxides and gold residues.
Cupellation is a technique that he uses in the final isolation of gold, silver or PGMs from the collector metal. At the scale of material handling Jason works with, a small cupel and a muffle furnace is all that is necessary for this step. Cupellation for gold isolation was described by Agricola in the 16th century. The lead collector mass selectively oxidizes to the PbO, or litharge, and diffuses into the cupel leaving behind the precious metal. Cupels were formerly made of bone ash or other materials that will not combine with the molten PbO to produce a viscous layer that would prevent seeping of the PbO into the container. This is also how gold was isolated in the old days by the assay office to determine the gold content of ore samples. Today several methods are available to assayers, including x-ray fluorescence.
For your viewing pleasure I have provided a link to a short but interesting video. It shows the disposal of large drums of wartime metallic sodium into a lake in Washington. It has that WWII news reel sound.
“Wartime sodium” in Washington suggests that the Na is from Hanford. Just a guess.
The term ‘bioorthogonal’ seems a little odd at first but the definition of orthogonal is- Adjective, intersecting or lying at right angles. This could stem from the idea of an atomic or molecular orbital whose axis is perpendicular to another resulting in bonding and anti-bonding orbital overlap. That is, they are electronically unconnected with each other. The analogy is with the fact that click reactions don’t interfere with biochemical reactions in the system. It’s a good choice of terms.
Sharpless achieved the vaunted adjective status prior to his earlier 2001 Nobel Prize in Chemistry for work on catalyzed chiral oxidations, i.e., the Sharpless Oxidation. To be an adjective in a named reaction in chemistry is the highest honor for a synthesis chemist.
With the appearance of COVID and polio the USA, the news has revealed that it is possible to detect and monitor certain viruses in municipal sewage. As a chemist I marvel at this. Sewage is a frightfully complex mixture of biological waste products along with many chemical cleaning products, detergents, grime and pharmaceuticals that go down the drain. How is it that one can collect enough intact virus particles from this fecal hell broth with enough purity to make a positive identification of genetic material?
A recent methodology is given in an article titled Detection of Pathogenic Viruses in Sewage Provided Early Warnings of Hepatitis A Virus and Norovirus Outbreaks and published in Appl Environ Microbiol. 2014 Nov; 80(21): 6771–6781, DOI: 10.1128/AEM.01981-14 by Maria Hellmér,aNicklas Paxéus,bLars Magnius,cLucica Enache,bBirgitta Arnholm,dAnnette Johansson,bTomas Bergström,a and Heléne Nordera,c. As you can see the work is from 2014 so this is not brand-spanking-new technology. It is interesting to note that the material used to sediment the viruses in this article was acidified powdered skim milk proteins. The article was found by a Google search and located at the NIH National Library of Medicine.
Why powdered skim milk? It could be that milk fat interferes with the process or the workers are just removing variables. More likely, it is because the widely available powdered milk that you buy at the grocery store is from skim milk. Dairy fat is too valuable for a business to squander and is used to make more profitable products like ice cream or whipping cream.
In the 2014 article above, the virus particles are extracted from the raw sewage onto acidified powdered skim milk proteins and amplified with quantitative polymerase chain reaction, qPCR. Powdered milk may seem strange but realize that virus particles can be removed by coagulation with metal ions, lime or with other polyelectrolytes, including proteins. The charge distribution on milk proteins will vary with acidity so these methods are very pH dependent. The viruses are naturally coated in proteins and thus will acquire surface charges varying with pH. The coagulation of proteins occurs when dissolved or suspended proteins irreversibly change their secondary structure by unfolding and condense to form a thicker solution or a solid form. The formation of cheese by acidification or solidifying a runny egg with heat are common examples of coagulation.
A 1973 review article by Gerald Berg in Bull World Health Organ. 1973; 49(5): 451–460, reviews methods for the removal of viruses from effluents, so knowledge of the sedimentation, or coagulation, of viruses in sewage has been around for a long while.
These articles are written by specialists in the field and may present considerable difficulty for a few readers. I would urge those so inclined to try to plow through the articles and pick up what you can. This holds true for all scientific papers. See what you can learn.